
Understanding The Regulatory Reality You're Actually Facing

Let's be honest, medical device regulation is tough. It's not just forms and boxes to check. It's a complex and ever-changing world of requirements, guidelines, and expectations. If you're not ready for it, it can seriously derail your medical device project, no matter how innovative it is. I've witnessed this firsthand, and trust me, it's not a pleasant sight. Companies invest their passion, energy, and significant funding into groundbreaking devices, only to be caught off guard by regulatory hurdles they never anticipated.
One of the most significant difficulties is the constant change. Old-school regulatory strategies often fall short because they're based on outdated rules. They don't account for the rapid progress in things like AI integration and cybersecurity, which are completely transforming the device approval process. Take, for example, software updates for AI-powered devices. The traditional model of infrequent updates just doesn't cut it when your algorithms are constantly learning and evolving. Regulators are still figuring this out, and companies need to be proactive in adapting their medical device regulatory strategy to keep up.
This dynamic environment has a big impact on the market. The global medical device regulatory affairs market is expected to hit USD 11.66 billion by 2030, growing at a CAGR of 9.55% between 2025 and 2030. This expansion is driven by advancements like the growing use of AI and machine learning in medical devices, along with increasingly stringent regulatory frameworks. Discover more insights. This means navigating this intricate regulatory landscape is crucial not just for thriving, but for survival.
The Hidden Costs Of Regulatory Missteps
Many companies don't fully grasp the consequences of regulatory missteps. It's not just about penalties and delays. A faulty regulatory strategy can damage investor trust, tarnish your brand's reputation, and ultimately kill a project, even one with massive potential. I recall working with a startup that had a game-changing device for early cancer detection. Their focus was so intensely on the technology that they overlooked the regulatory aspects. What happened? A delayed launch, frustrated investors, and a major company restructuring just to get back on track.
What The Smartest Players Are Doing Differently
The companies that flourish in this environment treat regulation not as an obstacle, but as a core element of their overall business strategy. They engage with regulatory bodies early and often, building relationships and getting advice from the beginning. They approach risk assessment as a strategic tool to inform their decisions, rather than a mere compliance exercise. They create flexible regulatory strategies that can adjust to the inevitable shifts along the way. They recognize that a proactive and adaptable medical device regulatory strategy isn’t just a cost of doing business; it’s a key competitive advantage. This change in perspective is vital for long-term success. This proactive approach enables them to anticipate hurdles, minimize disruptions, and ultimately bring their life-changing devices to market more quickly and effectively.
To illustrate some of the key differences in the global regulatory landscape, take a look at this comparison table:
Global Regulatory Framework Comparison
| Region | Approval Timeline | Key Requirements | Recent Changes |
|---|---|---|---|
| US (FDA) | Varies depending on device classification; can take several months to years. | Premarket approval (PMA) for Class III devices, 510(k) clearance for Class II, and general controls for Class I. Emphasis on clinical data and safety. | Increased focus on cybersecurity and AI/ML in medical devices. |
| EU (MDR) | Varies, generally longer timelines than previously under MDD. | Emphasis on clinical evidence, post-market surveillance, and unique device identification (UDI). More stringent requirements overall. | Transition from MDD to MDR has introduced significant new requirements and challenges for manufacturers. |
| Japan (PMDA) | Similar to FDA timelines. | Emphasis on quality management systems (QMS) and clinical trials conducted in Japan. | Ongoing efforts to harmonize regulations with international standards. |
| China (NMPA) | Can be lengthy and complex. | Registration and licensing procedures vary depending on the device classification. Emphasis on local testing and clinical data. | Increased scrutiny of foreign manufacturers and emphasis on local clinical trials. |
This table highlights just some of the key variations. As you can see, each region has its own nuances and priorities. Understanding these distinctions is critical when developing your regulatory strategy. You'll need to factor in these differences early on to avoid costly surprises down the line.
Building Your Foundation: Risk Assessment That Actually Drives Decisions

This screenshot from the FDA website highlights the criteria for classifying medical devices. It’s a good visual reminder that understanding your device’s classification is the very first step in a solid medical device regulatory strategy. The FDA uses a risk-based classification system. In a nutshell, the higher the risk to patients, the tougher the regulatory hoops you’ll have to jump through. This has a massive impact on your timeline and budget.
Let me give you an example. I worked with a company developing a Class III implantable device. They initially focused their risk assessment on the technical side – the device itself. They completely underestimated the clinical data the FDA would require and the inevitable back-and-forth that comes with regulatory submissions. They missed the regulatory risks altogether. That oversight cost them a year of delays and millions in unexpected expenses. Ouch. They learned a hard lesson: your medical device regulatory strategy has to account for all risks, not just the technical ones.
Identifying The Risks That Actually Matter
So many companies treat risk assessment like a checkbox exercise. They identify the generic risks everyone’s talking about at conferences and call it a day. But here’s the thing: the risks that really matter are the ones specific to your device and how it’s meant to be used. You have to go beyond the surface.
Here’s a practical framework I’ve used that actually helps drive decisions:
- Hazard Identification: Simple question, but vital: what could go wrong? Think about everything: material failures, software glitches, user errors… the whole nine yards.
- Risk Estimation: How likely is each hazard to actually happen, and what would the fallout be? Use data, talk to experts, look at similar devices – gather as much info as you can to inform your estimations.
- Risk Evaluation: Is the risk acceptable? This is where you compare the estimated risk to your pre-defined acceptance criteria. These criteria should align with regulatory guidelines and your own internal risk tolerance.
- Risk Control: If the risk isn't acceptable, how are you going to fix it? This is where you create your risk mitigation strategies – design changes, labeling updates, post-market surveillance plans, etc.
- Risk Documentation: Write it all down! Every single step. This isn’t just for ticking the compliance box. It’s a valuable tool for communicating with regulators and proving you’ve taken a proactive approach to patient safety.
Turning Risk Assessment Into A Competitive Advantage
A robust risk assessment does more than just keep the regulators happy. It can actually be a competitive advantage. By getting ahead of potential problems, you can streamline your development process, avoid costly delays, and build a safer, more reliable device. A well-defined medical device regulatory strategy, built on a rock-solid risk assessment, sends a clear message to investors and regulators: you take safety seriously. That translates into confidence and trust. It also helps attract talented regulatory professionals. Experienced folks want to work for companies that value their expertise and let them make a real difference in patient safety. Ultimately, this makes your company more attractive, speeds up the approval process, and sets you up for success in the competitive medical device market. Remember, getting your device to market efficiently and safely isn’t just about checking boxes. It’s about building a foundation for long-term growth and making a real impact.
Choosing Your Regulatory Pathway Like A Strategic Pro
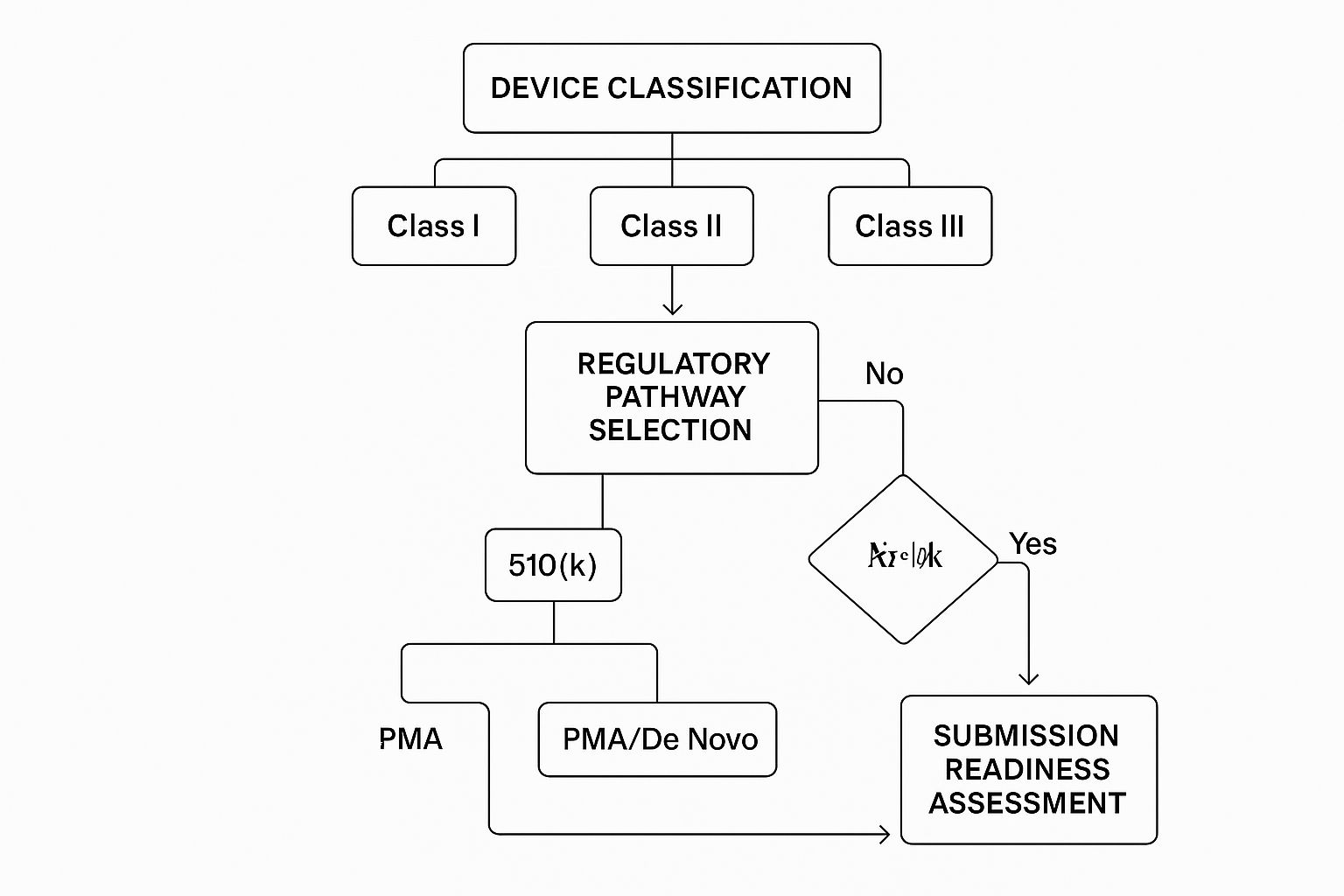
The infographic above gives you a visual overview of the key decisions involved in your medical device regulatory strategy. It shows how everything’s connected, starting with classifying your device and then moving through the different pathways—like 510(k), PMA, and De Novo—all the way to your final submission. You can see how each decision opens up different possibilities, highlighting just how important early planning and proper device classification really are. It can definitely feel a bit overwhelming, but trust me, with the right strategy, choosing a pathway can actually become one of your biggest advantages.
Knowing where the medical device market is headed is a critical piece of that strategy. The industry is projected to hit nearly USD 800 billion in annual sales, with consistent 5% yearly growth. This growth reflects the global need for innovative medical solutions. Learn more about future market trends. This booming market underscores why having a robust regulatory strategy is so important if you want to capitalize on these opportunities.
Matching Your Device to the Right Pathway
Choosing the wrong regulatory pathway is a bit like driving down a dead-end street. You end up wasting time, money, and forward momentum. I’ve seen companies go for the 510(k) because it seemed faster, only to discover their device was far too innovative to show substantial equivalence. They were forced to switch to a PMA, which cost them valuable time and significantly ramped up their development expenses. A good risk assessment framework relies on careful documentation, and setting up a reliable document control process is crucial for that. Meticulous documentation shows you understand your device's potential impact and keeps you aligned with regulatory requirements.
Let’s say you’re developing a new diagnostic device. If you can show it's substantially equivalent to an existing device, a 510(k) might be the right path. But if your device uses a brand-new technology or has a completely new intended use, a De Novo pathway might be a better fit, even though it's typically a lot more work. Understanding the specific requirements of each pathway is key.
Building Relationships, Not Just Submissions
Here’s a tip that often gets overlooked: build strong relationships with regulatory bodies. Early and frequent communication is essential. Don’t think of regulatory agencies as something to be feared. Instead, see them as partners who can help you get your device to market safely and efficiently. I've personally witnessed the benefits of pre-submission meetings with the FDA. Companies receive invaluable early feedback, helping them avoid common pitfalls and put together a submission package with a much better chance of approval.
Thinking Globally, Acting Strategically
If you envision your device having a global presence, think about international regulatory pathways right from the beginning. While parallel submissions can be more complex and costly, they can substantially shorten your time to market in various regions. This is particularly relevant if your device has strong market potential in multiple countries. Choosing the right pathway means understanding the dynamics of your target markets. For instance, the EU’s Medical Device Regulation (MDR) has significantly increased requirements for clinical evidence and post-market surveillance. Not accounting for these regional differences early on in your regulatory strategy can cause major problems later. Understanding these intricacies is crucial for creating a pathway strategy that optimizes your timeline and minimizes delays in getting your device to global markets.
Pivoting Without Panicking
Even with the best planning, sometimes your initial pathway choice just doesn't pan out. Don't let it throw you. Being adaptable is crucial in the world of medical device regulation. Learn from what didn’t work, gather new information, and be willing to change course. This could mean tweaking your clinical trial design, generating more data, or even looking at other pathways. The important thing is to stay flexible and persistent. Remember, the ultimate goal isn’t to rigidly stick to a plan, but to successfully bring your device to market while always prioritizing patient safety. A well-designed regulatory strategy should allow for adjustments and changes without completely derailing the process.
Mastering Submissions Without Losing Your Sanity

This screenshot from the FDA website, specifically their Premarket Notification 510(k) page, is a good example of the resources available for medical device manufacturers. It really highlights how important it is to be prepared and understand the submission process inside and out. Knowing where to find and how to use these resources is a huge part of getting your submission through.
Let's be honest, the mere mention of "submissions" can make even seasoned regulatory professionals cringe. They're intense. Late nights, endless document revisions, and that dreaded moment when reviewers ask for clarification on something you thought was crystal clear…it all comes with the territory. I've been there, trust me. I've even pulled my hair out over seemingly insignificant details that ended up becoming major roadblocks. But I've also learned a few tricks along the way that can help keep you sane and increase your chances of success.
Structuring Your Documents for Maximum Impact
Regulators are swamped with submissions. Your goal? Make their lives easier. Think of your submission as a story. It needs a compelling narrative, a logical flow, and supporting evidence that's easy to digest. Don't bury the crucial information. Make it shine. Use clear headings, concise language, and visuals wherever you can.
Here are a few tips from my experience:
-
Executive Summary: This is your elevator pitch. Make it concise and compelling, capturing the essence of your device, its intended use, and why it's worth approving.
-
Logical Organization: A consistent structure is your best friend. Reviewers need to find what they're looking for quickly and easily.
-
Anticipate Questions: Put yourself in the reviewer's shoes. What questions would you ask? Addressing these proactively saves everyone time and headaches.
The Art of Regulatory Communication
Effective communication is key. Responding to information requests isn't just about answering questions; it's about building a relationship with the reviewers and showing them you understand the process. Ditch the jargon. Be clear, concise, and responsive. Choosing the right regulatory pathway often involves understanding different compliance frameworks. If you need a refresher on that, check out this helpful article on choosing your compliance framework.
Here’s what I’ve found works best:
-
Respond Promptly: Don’t leave reviewers hanging. A quick turnaround demonstrates that you’re attentive and organized.
-
Be Thorough, But Concise: Answer everything completely, but don't overload them with unnecessary information. Get to the point.
-
Be Professional and Collaborative: Treat the reviewers as partners. A positive working relationship can make a big difference.
Speaking of communication, let's talk about the ever-changing regulatory landscape. In 2025, we're seeing increased challenges due to global harmonization, AI guidelines, and tighter cybersecurity requirements. In the U.S., recalls have hit a four-year high, with Class I recalls at a 15-year peak. This highlights how crucial a solid regulatory strategy really is. This article provides even more insights into the current environment and why adaptability is so important.
Managing Internal and External Stakeholders
Submissions often involve a delicate balancing act between internal teams who need updates and external consultants who can sometimes overcomplicate things. Clear expectations and communication channels are essential from the very beginning.
Here are some things I've learned:
-
Regular Communication: Keep everyone in the loop with regular updates, but don’t bombard them with unnecessary emails. Find a balance.
-
Project Management Tools: These are invaluable for tracking progress, managing documents, and making sure everyone’s on the same page. I've found Asana and Trello to be particularly helpful.
-
Don't Be Afraid to Push Back: If a consultant’s advice doesn’t sit right, question it. You're the expert on your device.
Building Realistic Timelines
Delays are inevitable. Factor them into your timeline from the get-go. I can't tell you how many projects I've seen go off the rails because of overly optimistic timelines that didn't account for the inevitable back-and-forth with regulators. Add a buffer to each stage of the process. This allows you to handle unexpected issues without derailing the whole project. A realistic timeline also helps manage internal expectations and keeps everyone focused on the bigger picture of your overall regulatory strategy.
To help you visualize timelines, I've put together this table:
Submission Timeline and Requirements Matrix
Expected timelines and key requirements for different regulatory pathways
| Pathway | Average Timeline | Key Documents | Success Rate |
|---|---|---|---|
| 510(k) | 90-180 days | Substantial Equivalence Report, Device Description, Labeling | 85% |
| PMA | 180-365+ days | Clinical Trial Data, Device Description, Manufacturing Information | 70% |
| De Novo | 180-270 days | Benefit-Risk Analysis, Device Description, Proposed Special Controls | 60% |
Note: These timelines and success rates are averages and can vary depending on the specifics of each device and submission.
Remember, each submission is a learning opportunity. Reflect on what went well, what could have been improved, and how to refine your process for the next one. This iterative approach to constantly improving your regulatory strategy is crucial for navigating the complex world of medical devices and achieving long-term success.
Post-Market Strategy: Staying Compliant When It Really Counts
So, you got your medical device approved. Huge congrats! Seriously, that’s a massive accomplishment. But, and I hate to be the bearer of slightly less-good news, it's really just the beginning. The real test of your medical device regulatory strategy starts after the launch.
This is where things get real. You're dealing with actual patient data, unexpected hiccups, and regulations that sometimes feel like they change by the minute. I've seen companies pop the champagne after getting approval, only to trip and fall post-market because they weren't ready for what comes next. Their regulatory strategy didn’t look past the approval finish line, and that mistake cost them big time.
This snapshot from the FDA website lays out the various post-market requirements. One look and it’s obvious: post-market surveillance isn't a one-and-done deal. It’s ongoing, continuous reporting, analysis, and sometimes taking action to make sure your device stays safe and effective. Knowing how to navigate this is key to keeping your device on the market and building trust with patients and doctors.
Building a Sustainable Post-Market Surveillance System
Post-market surveillance isn’t about checking boxes. It’s about learning from real-world use and constantly making your device better. It's about catching problems before they blow up, adapting to new info, and showing everyone you're serious about patient safety. The trick is building a system that works without costing a fortune.
Here’s what I’ve picked up from companies who’ve successfully navigated this:
-
Proactive Data Collection: Don’t wait for fires to start. Actively collect data on how your device is performing, get user feedback, and keep an eye out for potential adverse events. This helps you spot trends and address any safety concerns before they snowball.
-
Automated Reporting: Tech is your friend here. Automated systems help make sure you’re reporting adverse events and other crucial data to regulatory bodies accurately and on time.
-
Regular Analysis: Data collection is only half the battle. You have to analyze it! Regularly review your post-market data to identify trends, catch potential safety signals, and pinpoint areas for improvement. This is how you make smart decisions about product tweaks, label updates, or even recalls.
Handling Adverse Event Reporting Without Panicking
Adverse events are a fact of life. It's inevitable. It’s how you deal with them that counts. A good process can stop a small problem from becoming a five-alarm fire.
Here's the deal:
-
Have a Plan: You need a clear, step-by-step plan for reporting adverse events. Who does what? What are the communication channels? Get it all mapped out.
-
Be Transparent: Don’t try to sweep adverse events under the rug. Transparency builds trust with regulators and patients.
-
Learn From Every Event: Every adverse event is a chance to learn and improve. Use that information to make your device, your processes, and your overall regulatory strategy stronger.
Staying Ahead of Regulatory Changes
Regulations change. A lot. Staying on top of these changes and adjusting your strategy is critical. This means keeping a close eye on regulatory updates, going to industry events, and being willing to change your plans when you need to. Companies that set up internal processes to track and understand these changes are much better prepared and avoid expensive surprises. After market approval, a solid business compliance checklist is your best friend for ensuring you stay compliant.
Turning Post-Market Data into Competitive Advantage
Post-market data is a gold mine. Use it to shape your next device, refine your regulatory strategy, and gain a real competitive edge. Companies that effectively use this data can discover unmet needs, improve their products, and create devices that are safer, more effective, and better tailored to patients and healthcare professionals. It can even help streamline future regulatory submissions by giving you real-world evidence of your device's performance and safety. This creates a competitive advantage that's hard to beat.
Remember, post-market surveillance isn't a headache, it’s an opportunity. By seeing it as a strategic tool, you can strengthen your market position, build trust, and improve patient outcomes.
Building Your Regulatory Dream Team That Actually Delivers
Building a successful medical device regulatory strategy is a team effort. It’s not a solo mission. I’ve seen firsthand how the right team can make regulatory submissions feel almost effortless, while a dysfunctional team can turn even simple submissions into a nightmare.
Internal Expertise Vs. External Consultants: Finding the Right Balance
One of the biggest questions you’ll face is deciding between building an internal regulatory team or outsourcing to consultants. There’s no magic bullet answer here. Startups often begin with consultants to tap into that readily available experience while they build internal capabilities. Larger companies might have full-fledged regulatory departments, but still use specialized consultants for particularly tricky projects. I’ve seen companies try to save money by keeping everything in-house, only to discover they were missing key expertise for certain submissions. In the end, that cost them more time and money than if they had just hired a consultant from the get-go.
Identifying Top Regulatory Talent
Finding the right people makes all the difference. Look for individuals with a solid grasp of the regulations, excellent communication skills, and a proactive approach. They need to be able to see potential problems coming and work well with other teams within the company. I’ve worked with regulatory professionals who were incredibly knowledgeable, but just couldn't explain things clearly to reviewers or internal stakeholders. That kind of communication breakdown can create major roadblocks.
Evaluating Regulatory Consultants: Avoiding Costly Mistakes
If you decide to work with consultants, do your homework. Don’t just pick the cheapest option or the one with the slickest presentation. Look for consultants with a proven history of success in your specific device area. Ask for references and look at case studies. I've seen companies choose consultants who overpromised and underdelivered, resulting in expensive delays and rework. It’s a painful lesson to learn.
Building Institutional Knowledge and Streamlining Workflows
As your team grows, you absolutely have to create a system for documenting and sharing regulatory knowledge. Things like documented processes, templates, and clear communication channels can ensure consistency and prevent everyone from having to reinvent the wheel with each submission. I’ve seen companies lose precious time and expertise when someone leaves and takes all that knowledge with them. After market approval, maintaining compliance is vital; a solid business compliance checklist is essential.
Budgeting for Regulatory Talent: Getting the Most Bang for Your Buck
Regulatory affairs can be pricey. But think of it as an investment, not a cost. A realistic budget is essential. Include salaries, consultant fees, training, and software tools in your calculations. I’ve seen companies underestimate their regulatory budget, which just leads to scrambling for funds later and inevitable project delays.
Measuring Team Performance: Beyond Checkboxes
Evaluating your regulatory team's performance is about more than just counting submissions. Look at things like approval timelines, the number of questions you get from reviewers, and your post-market compliance record. Those metrics give you a much better idea of how your team is contributing to your overall business goals.
Creating a Culture of Regulatory Excellence
A strong regulatory culture isn’t just about hiring the right people. It’s about cultivating an environment of proactive compliance, continuous learning, and open communication. This makes regulatory professionals feel valued and empowered. I’ve seen companies transform their regulatory function from a roadblock into a strategic advantage by building a culture that values regulatory excellence. This not only strengthens compliance but also attracts and retains top talent.
Your Regulatory Strategy Action Plan That Actually Works
Okay, so we’ve talked a lot about the moving parts of a solid regulatory strategy. Now, let's get down to brass tacks. How do you take all this information and build a medical device regulatory strategy that actually works? This isn’t just a recap; think of it as your personalized action plan. It boils down to honestly assessing your current regulatory situation, finding the gaps that truly matter, and making smart improvements that deliver maximum impact without blowing your budget.
Honestly Assessing Your Current Regulatory Position
First, you need a brutally honest assessment of where you stand. Are you treating regulatory requirements as an afterthought, something you’ll “get to later,” or is it woven into the fabric of your business strategy? Be honest. I’ve seen companies with game-changing devices stumble, not because the device was flawed, but because they underestimated the importance of regulatory planning. They saw it as a hurdle to jump over, not a fundamental component of success.
Here’s a simple checklist to get you started:
-
Documented Regulatory Strategy: Do you even have one written down? If not, that’s priority number one.
-
Risk Assessment: Is yours thorough and specific to your device, or is it a generic, one-size-fits-all document?
-
Regulatory Expertise: Do you have the right people on your team or access to external experts who can guide you?
-
Post-Market Plan: What happens after your device launches? Do you have a system in place for monitoring performance and handling any issues that arise?
Identifying and Prioritizing Gaps
Once you’ve taken stock of your current situation, pinpoint the gaps. Where are you falling short? What needs work? Don’t just list them out—prioritize them. Which gaps pose the biggest threat to your project, and which ones offer the greatest potential for improvement? I’ve worked with companies that tried to fix everything at once, spreading their resources too thin and ultimately accomplishing very little. Focus on the gaps that truly matter.
Building Realistic Timelines and Budgets
Now, let’s talk timelines. Be realistic. Regulatory processes always take longer than you anticipate. Build in buffer time. Trust me on this one. And while we’re at it, let’s not forget the budget. Regulatory affairs cost money. Factor in expenses for consultants, testing, and everything else. Underestimating your regulatory budget can seriously derail a project later on.
Establishing Meaningful Metrics
How will you measure the effectiveness of your medical device regulatory strategy? You need metrics. Think about things like time to approval, the number of adverse events reported, or your post-market compliance rates. Track these metrics and use the data to fine-tune your strategy as you go.
Building a Scalable and Adaptable Strategy
The regulatory world is constantly evolving. Your strategy needs to be flexible enough to adapt to these changes. This means staying on top of regulatory updates, attending industry conferences, and being willing to adjust your course when necessary. I've seen companies stick to outdated strategies, only to be blindsided by new regulations. Don’t let that be you.
Practical Checklists, Warning Signs, and Success Indicators
For each phase of your regulatory journey, develop clear checklists. What needs to be done? When? Who's responsible? Also, identify potential warning signs that could signal trouble ahead—things like missed deadlines, unexpected questions from regulators, or negative user feedback. Finally, define what success looks like for each phase.
Building a Culture of Regulatory Proactiveness
Last but not least, build a company culture where everyone understands the importance of regulatory compliance. This isn't just the regulatory team's responsibility; it's everyone's. When everyone is on board, regulatory affairs becomes an integral part of your company’s DNA, not a dreaded chore.
By following these steps, you can build a medical device regulatory strategy that doesn't just tick the compliance boxes but actively helps you bring your device to market efficiently and effectively.
Ready to integrate AI into your medical imaging process and navigate the regulatory landscape with confidence? PYCAD offers expert support from data handling and model training to deployment and post-market compliance. Learn more about how PYCAD can help you.