The world of medical devices runs on a non-negotiable principle: safety first. The regulatory requirements for medical devices are the legal bedrock that ensures this, outlining the specific obligations manufacturers must meet before a product ever reaches a patient. This isn't just bureaucratic red tape; it's a comprehensive system covering everything from quality management and clinical evidence to post-market surveillance.
These rules exist to make sure every device—whether it's a simple tongue depressor or a sophisticated AI-powered diagnostic tool—is proven safe and effective for its intended use.
Why Medical Device Regulation Is More Critical Than Ever

Think of regulatory approval as a global passport for your medical device. Without that essential stamp from an authority like the U.S. Food and Drug Administration (FDA) or an EU Notified Body, your product is stuck at the border. It simply can't be sold or used on the patients who need it most.
This "passport" is far more than a piece of paper. It's a testament that your device has been through the wringer—rigorously vetted and validated. And getting that approval has never been more challenging. Technology is moving at lightning speed, with innovations in software, AI, and miniaturization constantly redefining what a medical device is and what it can do. Regulators are in a constant race to keep up, creating a highly dynamic environment where the rules are always evolving.
The Rising Stakes in a High-Tech World
The sheer size of the global medical device regulatory affairs market tells the story. It's projected to hit USD 11.66 billion by 2030, growing at a compound annual growth rate (CAGR) of 9.55%. This explosive growth isn't just about more paperwork. It’s a direct reflection of stricter global standards, the complex needs of AI-driven devices, and the desperate demand for specialized regulatory expertise.
This high-stakes environment means a rock-solid compliance strategy is no longer optional—it's a foundational pillar of your business plan. It's not something to tack on at the end but a core component for successful market entry. The entire innovation pipeline, and the regulatory demands that come with it, is also deeply tied to the financial support available for basic science. Major shifts in funding for biomedical research can have a ripple effect on the types of new devices developed and brought to market.
A well-executed regulatory strategy is no longer just about avoiding fines. It is a critical business enabler that builds trust with patients, clinicians, and investors, ultimately unlocking market access and creating a sustainable competitive advantage.
Key Gatekeepers in the Global Arena
To truly get a handle on medical device regulations, you need to know the major players. Every significant market has its own "gatekeeper," a regulatory body with a clear mission to protect public health. The table below offers a quick snapshot of the key authorities you'll encounter.
Major Global Medical Device Regulatory Authorities at a Glance
| Regulatory Body / Framework | Region | Primary Responsibility |
|---|---|---|
| Food and Drug Administration (FDA) | United States | Oversees device approval using a risk-based classification system (Class I, II, III). |
| EU MDR / IVDR | European Union | Sets the rules for medical devices (MDR) and in-vitro diagnostics (IVDR), with Notified Bodies assessing conformity. |
| Health Canada | Canada | Regulates the safety, effectiveness, and quality of medical devices sold in Canada. |
| PMDA | Japan | The Pharmaceuticals and Medical Devices Agency reviews and approves medical devices and drugs for the Japanese market. |
| Therapeutic Goods Administration (TGA) | Australia | Ensures medical devices available in Australia are safe and fit for their intended purpose. |
While this list isn't exhaustive, it highlights the main authorities you'll need to work with. Each one has its own unique framework and set of hoops to jump through, which is the core challenge for any device manufacturer with global ambitions.
This guide will serve as your map. Now that we've covered the stakes and the "why" behind the rules, we can dive into the specific steps you'll need to take to achieve compliance.
The Four Pillars of Medical Device Compliance
If you're trying to make sense of global regulations, it helps to start with the fundamentals. Think of it like building a house. You wouldn't just start putting up walls without a solid foundation, a detailed blueprint, an engineer's structural report, and a user manual for the new owner. The regulatory requirements for medical devices are built on a very similar set of core pillars.
Once you get a handle on these four concepts, you'll have a framework that works whether you're aiming for the US, the EU, or another major market. They are the essential, interconnected parts that prove your device is stable, safe, and ready for patients.
Pillar 1: Quality Management System (The Foundation)
Your Quality Management System (QMS) is the absolute bedrock of your entire compliance strategy. It's the documented collection of all your processes, procedures, and responsibilities for making sure your device meets its quality goals. A QMS isn't just a binder collecting dust on a shelf; it's the living, breathing rulebook for how your company operates.
It governs everything from the first design sketch and choosing suppliers to manufacturing controls and handling customer complaints. The international standard ISO 13485:2016 is the globally accepted framework for a medical device QMS. In fact, the US FDA is now harmonizing its own quality system rules with this standard through the Quality Management System Regulation (QMSR) to create more consistency worldwide.
Simply put, a strong QMS gives you consistency and control. It’s the difference between building on solid concrete versus shifting sand—it provides the stability needed for every other compliance activity you undertake.
Pillar 2: Device Classification (The Building Code)
Not all medical devices are the same, so the rules they have to follow aren't the same either. Device Classification is how you categorize your product based on its intended use and the level of risk it poses to the patient or user. This classification is what determines the specific "building code"—or regulatory pathway—you'll need to follow.
This diagram illustrates the risk-based classification system used by most regulatory bodies across the globe.
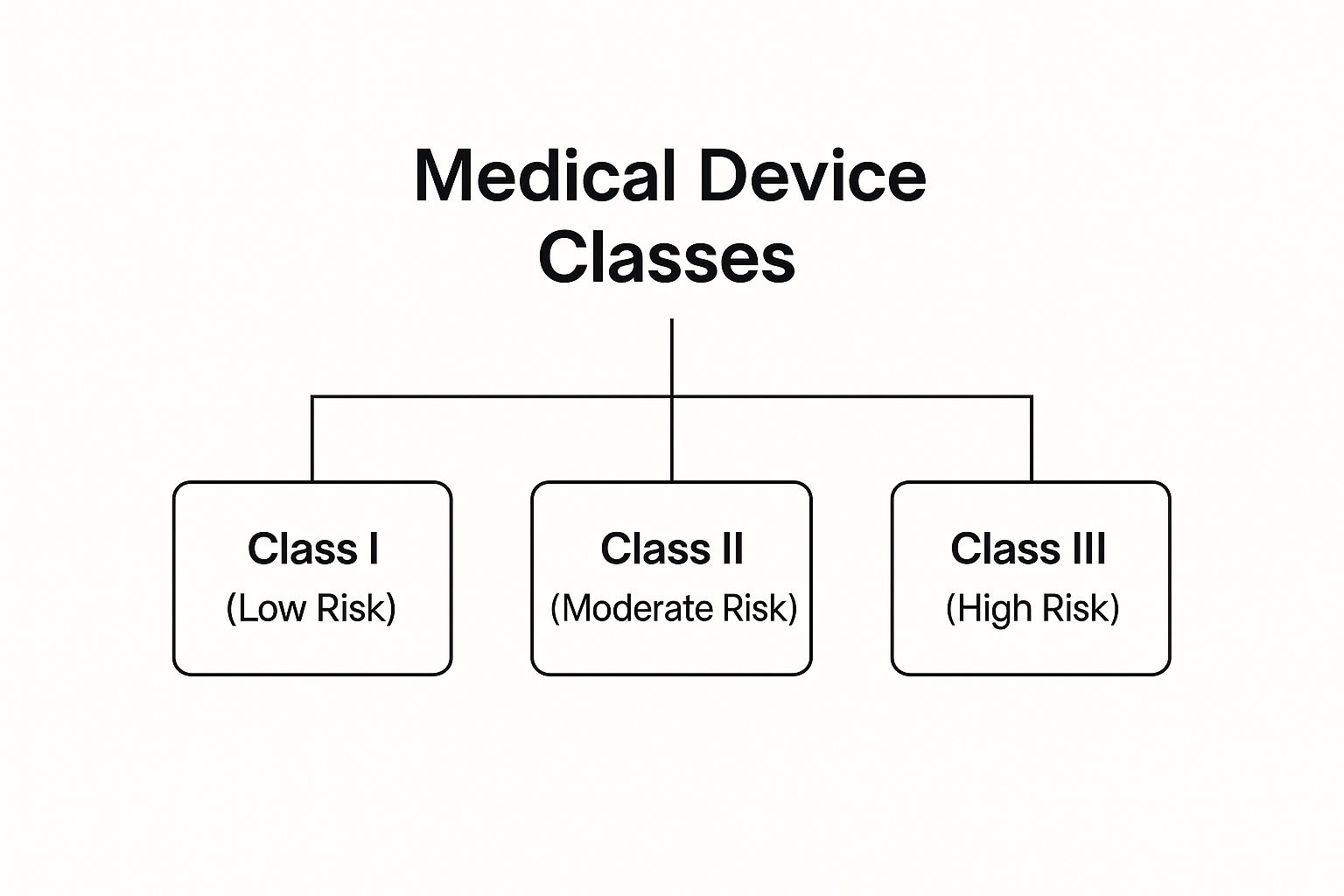
As you can see, when a device’s risk level increases from Class I to Class III, the amount of regulatory scrutiny and required proof goes up dramatically.
A low-risk device like an elastic bandage (Class I) needs far less oversight than a high-risk, life-sustaining device like an implantable pacemaker (Class III). Getting the classification wrong is one of the most common—and costly—early mistakes a company can make. It sends you down a completely wrong path, wasting a huge amount of time and money.
Pillar 3: Clinical Evidence (The Structural Report)
If classification is your blueprint, then clinical evidence is the structural engineer's report that proves your house is safe to live in. It's the body of clinical data and performance evaluations you gather to show that your device actually does what you say it does, and that its benefits outweigh any potential risks.
Clinical evidence isn't about hoping your device works; it's about proving it works. This objective proof must be scientifically valid and sufficient to support your claims.
The amount of clinical evidence you need directly corresponds to the device's risk class:
- Low-Risk Devices: For these, you can often gather evidence from existing scientific literature on similar, well-established technologies.
- Moderate-Risk Devices: This usually requires a combination of literature reviews, bench testing, and sometimes data from clinical studies.
- High-Risk Devices: These almost always demand extensive, formal clinical investigations (human trials) to collect solid data on safety and efficacy before they can even be considered for approval.
This pillar is all about generating indisputable proof that your device is both safe and effective for its specific purpose.
Pillar 4: Labeling and Post-Market Surveillance (The User Manual and Ongoing Maintenance)
Finally, even the best-built house needs a clear user manual and a plan for long-term upkeep. In the device world, labeling is your user manual. This includes all the information provided to the user, from the text on the box and the physical label to the detailed Instructions for Use (IFU). It has to be clear, accurate, and tell clinicians and patients exactly how to use the device safely and effectively.
But your job isn't done at launch. Post-Market Surveillance (PMS) is the "ongoing maintenance" plan for your device. It’s a proactive system you must have in place to monitor the device's performance out in the real world, collect feedback, and report any adverse events. This ensures that any unexpected problems are identified quickly, protecting patients long after your product is on the market.
Mastering the US FDA Regulatory Pathways

Trying to get a medical device to market in the United States can feel a lot like trying to open a high-security vault. It seems impenetrable from the outside, but success really comes down to having the right key for the right lock. The regulatory requirements for medical devices in the US are built around a few primary "keys," or pathways, each designed for a specific level of device risk.
The key you'll need is determined almost entirely by your device's classification. For low-risk Class I and most moderate-risk Class II devices, the go-to path is the Premarket Notification, far better known as the 510(k). For high-risk Class III devices—the ones that sustain or support life—the journey is through the rigorous Premarket Approval (PMA) pathway. And for truly novel low-to-moderate risk devices that don't have a clear equivalent, the De Novo pathway offers a third route to market.
The 510(k) Pathway Demystified
The vast majority of devices find their way into the U.S. market through the 510(k) process. This is the most common route because it doesn't force you to prove absolute safety and effectiveness from square one. Instead, your main job is to demonstrate that your new device is substantially equivalent to a legally marketed device that's already out there.
This existing device is called a "predicate device." Think of it like a trusted character witness in a trial. Your goal is to convince the FDA that your device is so similar to this "witness"—in how it's used, its technology, and its performance—that it doesn't introduce any new or different safety and efficacy questions.
To make your case, you'll need to compile a comprehensive file that includes:
- Detailed Device Description: A clear explanation of what your device is, its components, and exactly how it functions.
- Comparison to Predicate: A meticulous, point-by-point analysis showing how your device measures up against the predicate in design, materials, and purpose.
- Performance Testing Data: This includes bench testing and, in some cases, animal or clinical data that backs up your claims of equivalence.
The FDA reviews this submission to decide if your device is, in fact, substantially equivalent. If they agree, they issue a clearance letter, and you're officially cleared to market your device in the US.
The High Bar of Premarket Approval
The Premarket Approval (PMA) pathway is the FDA's most demanding review process, and it's reserved for Class III devices. This makes sense, as these devices—think pacemakers or artificial heart valves—carry the greatest potential risk to patients. Unlike the 510(k), a PMA isn't about finding a similar device to compare yours to.
A PMA requires you to provide valid scientific evidence proving your device is safe and effective for its intended use. There is no shortcut through substantial equivalence; your device must stand on its own merit.
This means you’re on the hook for conducting extensive clinical trials to gather your own robust data. The PMA application itself is a monumental task, often running thousands of pages filled with technical, pre-clinical, and clinical information. The review is just as intensive, involving a deep scientific and regulatory dive by FDA experts. Approval is only granted when the FDA determines that the device's benefits to health decisively outweigh its known risks.
The Evolving Role of Software and Cybersecurity
In recent years, the FDA has put software and cybersecurity under a microscope, fundamentally changing the regulatory requirements for medical devices. This is especially true for Software as a Medical Device (SaMD) and any connected device that can be accessed remotely. The FDA finalized guidance in 2022 that now requires electronic 510(k) submissions, a clear move away from older, less secure formats.
This shift means that simply providing basic performance data is no longer enough. Your submission must now include detailed documentation on:
- Software Validation: Concrete proof that your software performs as intended, reliably and consistently.
- Cybersecurity Risk Management: A complete analysis of potential cybersecurity vulnerabilities, from unauthorized access to malware attacks.
- A "Software Bill of Materials" (SBOM): A comprehensive list of all third-party software components in your device, which helps flag potential security risks hidden in the supply chain.
Failing to properly address cybersecurity has become a common reason for submission delays and outright rejections. The FDA now expects you to build security into your devices from the ground up, not bolt it on as an afterthought. This proactive mindset is critical for gaining market clearance and, more importantly, for protecting patient safety in our increasingly connected world.
If you thought the US FDA process was complex, get ready for a whole different ball game in Europe. Gaining access to the European market means mastering a new, far more stringent security system: the Medical Device Regulation (MDR) and the In Vitro Diagnostic Regulation (IVDR).
This isn't just a simple update to the old rules. It's a ground-up overhaul designed to put patient safety and transparency first. Think of it like swapping out your basic home alarm for a fortified, continuously monitored bank vault. The goal is the same—security—but the intensity and diligence required are on another level entirely. For manufacturers, this means the old playbook is obsolete. You have to adapt to a much tougher framework to earn and keep that all-important CE Mark, your non-negotiable ticket to sell in Europe.
The Higher Standard for Clinical Evidence
One of the biggest hurdles under MDR and IVDR is the much stricter demand for solid clinical evidence. Under the old Medical Devices Directive (MDD), companies could often lean on data from "equivalent" devices to prove their product was safe and effective. That loophole is now almost completely closed.
While the idea of equivalence technically still exists, the criteria are so tight that it's incredibly difficult to prove. The practical result? You're now expected to generate your own clinical data for your specific device, especially if it falls into a higher-risk category.
The new regulations are built on a simple but demanding principle: If you make a claim, you have to back it up—with your own data. The burden of proof has shifted entirely to the manufacturer to demonstrate safety and performance not just at launch, but throughout the device's entire lifecycle.
This lifecycle approach means your job is never really done. You are now required to have a proactive Post-Market Clinical Follow-up (PMCF) plan. This means you must actively gather and analyze data on how your device performs in the real world to continuously confirm that its benefits outweigh any risks.
EUDAMED and the Push for Total Traceability
Another major pillar of the new European framework is the drive for complete transparency and traceability. The engine behind this is a massive centralized system: the European Database on Medical Devices (EUDAMED). While its full rollout has been phased, its purpose is crystal clear.
Once fully operational, EUDAMED will be a public-facing, one-stop shop for information on:
- Device Registration: Every single device must be registered with a Unique Device Identifier (UDI).
- Economic Operators: All manufacturers, authorized representatives, and importers have to register themselves.
- Notified Bodies and Certificates: Every certificate issued will be logged and available for review.
- Clinical Investigations: Key details about ongoing and completed clinical studies will be public.
- Vigilance and Post-Market Surveillance: All reports on serious incidents and safety actions will be accessible.
This central database effectively puts the entire industry inside a glass box. Regulators, doctors, and even patients can access crucial data, creating a powerful tool for market surveillance and ensuring every device can be tracked from factory to patient.
New Roles and Responsibilities
The MDR also created a brand-new, mandatory role within every medical device company: the Person Responsible for Regulatory Compliance (PRRC). This isn't just a title; it's a designated individual or team with proven expertise who is legally on the hook for overseeing key compliance activities.
The PRRC’s core duties include making sure that:
- A device’s conformity is properly checked before it’s released to the market.
- All technical documentation and the EU declaration of conformity are always up-to-date.
- Post-market surveillance and reporting obligations are fully met.
- For investigational devices, the proper statement of compliance is issued.
This requirement forces accountability directly into the organizational chart. You must have a designated expert whose sole job is to guarantee your compliance systems are not just designed correctly, but are working perfectly day in and day out. From clinical proof and supply chain tracking to post-market vigilance, the EU's MDR and IVDR frameworks have undeniably raised the bar. To see how these changes compare to other global systems, you can explore this detailed compliance roadmap.
Future-Proofing Your Compliance Strategy

In the world of medical device regulation, standing still means falling behind. Just clearing today's compliance hurdles isn't enough. The real challenge—and the greatest competitive advantage—lies in building a compliance framework that anticipates what's coming next. A forward-thinking strategy allows you to adapt to regulatory shifts proactively, not just react when they happen.
This forward-looking mindset is critical because the regulatory requirements for medical devices are constantly evolving. Around the globe, regulators are racing to keep up with disruptive technologies and new patient risks, which means the ground is always shifting beneath our feet. To build a truly resilient strategy, you need to spot these trends early and weave them into your planning today.
The Rise of AI and Software as a Medical Device
Two of the biggest game-changers are Artificial Intelligence (AI) and Software as a Medical Device (SaMD). These aren't just futuristic concepts anymore; they are here, and they bring entirely new validation challenges to the table. A physical instrument has fixed, predictable parts. An AI model, on the other hand, can learn, adapt, and change over time, which completely upends traditional approval models designed for static products.
This leaves regulators wrestling with some tough questions. How can you properly validate an algorithm that is designed to evolve? What kind of documentation can prove its decisions are transparent and free from bias?
The core challenge with AI-driven devices is ensuring their safety and effectiveness throughout their entire lifecycle, not just at the moment of approval. This requires a shift from a one-time validation to a continuous monitoring and re-validation process.
As a manufacturer, you should prepare for much deeper scrutiny in several key areas:
- Algorithm Transparency: You'll need to clearly explain how your AI model works and what data it was trained on. "Black box" explanations won't cut it.
- Change Control Protocols: Regulators will want to see a rock-solid plan for how you'll manage, validate, and roll out software updates or algorithm changes after the product is already on the market.
- Bias Detection and Mitigation: It's becoming essential to prove that the AI performs equitably and accurately across diverse patient populations.
The New Frontier of Cybersecurity and Sustainability
Beyond AI, two other powerful trends are reshaping what compliance looks like: cybersecurity and environmental sustainability.
The explosion of connected, "smart" devices has elevated cybersecurity from an IT issue to a critical patient safety concern. A security breach is no longer just a data privacy problem; it can directly compromise a device's function and lead to real patient harm. As a result, new cybersecurity mandates are rolling out globally, including the EU’s Cyber Resilience Act and proposals in the US.
At the same time, the push for sustainability in healthcare is gaining serious momentum. Regulators are starting to examine the entire lifecycle of a device. They're asking about everything from eco-friendly design principles that reduce environmental impact to the responsible management of single-use plastics. Companies that get ahead of this by integrating sustainable practices into their design and manufacturing will find themselves much better positioned for the regulations of tomorrow.
Keeping up with these changes is a massive undertaking. To stay ahead of the curve, many organizations are turning to advanced tools, like an AI Healthcare Policy Analyzer, to help decipher complex regulatory documents and forecast future requirements. Taking this proactive approach is no longer a luxury—it’s the key to navigating what comes next.
Common Regulatory Pitfalls and How to Avoid Them
Getting a medical device to market can feel like navigating a minefield. You can have the best plan in the world, but success often hinges on knowing where the traps are buried. The quickest way to avoid costly delays and blown budgets is to learn from the missteps of others.
Three particular pitfalls consistently trip up even the most innovative medical device companies. These aren't obscure legal loopholes; they're fundamental mistakes in strategy that can sink a project. Understanding them is the first step toward a much smoother path to market.
Misclassifying Your Device Early On
One of the most common—and damaging—mistakes is getting your device classification wrong. This one decision, made right at the start, dictates your entire regulatory journey. It sets the requirements for your documentation, clinical data, and overall costs.
Think of it like choosing the wrong foundation for a house. If you build a skyscraper on a foundation meant for a shed, the entire structure is compromised from day one.
Imagine a startup with a new diagnostic app. They assume it's a low-risk Class I device and move forward with minimal oversight. Months later, regulators inform them that the app's AI-powered predictions actually place it in the higher-risk Class IIa category. Suddenly, their timeline is pushed back by a year, their budget is shot, and they have to start over to gather the right evidence.
How to avoid this:
- Get Expert Advice Early: Talk to a regulatory consultant before you build a prototype or write a single line of code. Don't guess.
- Document Your Reasoning: Don't just pick a class. Write down exactly why you chose it, referencing the specific classification rules, your device's intended use, and its mechanism of action.
- Plan for the Higher Risk: If your device falls into a grey area between two classes, always prepare for the more stringent one. It's much better to be overprepared than to be sent back to the drawing board.
Submitting an Incomplete Technical File
Another huge stumbling block is an incomplete technical file or 510(k) submission. Regulators need a crystal-clear, comprehensive story that proves your device is safe and performs as intended. If you submit a file with missing data, weak justifications, or a shoddy risk analysis, you’re guaranteeing yourself a rejection or a long, painful back-and-forth.
Your submission isn't a data dump—it's a persuasive argument. Every single claim you make must be backed up by solid, traceable evidence. An incomplete file tells regulators you haven't done your homework, and their trust in your product plummets.
These rejections often happen because of simple but critical oversights, like poor software validation or a flimsy cybersecurity plan. These issues aren't just paperwork problems; they are leading causes of recalls. In the U.S., recall figures recently hit a four-year high, with the most severe Class I recalls reaching a 15-year peak. The top reasons for these recalls worldwide? Software failures and other technical device flaws. You can get a much deeper look at these statistics by reading the full medical device recall report.
Underestimating Post-Market Responsibilities
Finally, too many companies see regulatory approval as the finish line. In reality, it’s the starting gun. The regulatory requirements for medical devices don't end at launch; they cover the entire life of your product.
Ignoring your post-market surveillance (PMS) and vigilance reporting duties isn't just a bad look—it's against the law. If you fail to collect data on how your device works in the real world or don't report adverse events, you're risking massive fines, forced recalls, and losing your permission to sell the device at all. This is about more than just checking a box; it's about your ongoing duty to keep patients safe long after you've made the sale.
Your Medical Device Regulation Questions Answered
Let's be honest: trying to untangle the web of regulatory requirements for medical devices can feel like a nightmare. You're not alone in feeling this way. A recent survey found that a staggering 72% of life sciences executives see regulatory compliance as one of their top three business hurdles. If you want to dig deeper, you can learn more about these industry challenges in the full report.
To cut through some of that noise, we’ve put together answers to the real-world questions we hear all the time. We'll cover where to begin, how markets differ, and what compliance looks like long after your device hits the shelves.
What Is the First Step for a Startup?
So, you have a groundbreaking idea for a medical device. What now? Before you do anything else, you must formally classify your device. This isn't just paperwork; it’s the foundational step that defines your entire journey.
Your device's classification is determined by its intended use and the level of risk it presents to a patient. This single decision dictates everything that follows—the documentation you'll need, the clinical evidence you have to gather, and your overall timeline and budget. Getting this wrong can set you back months, if not years, and drain your resources. Bringing in a regulatory expert at this stage isn't an expense; it's an investment to prevent costly mistakes down the road.
Is FDA Approval Valid in Europe?
This is a common point of confusion, and the answer is a firm no. Regulatory approvals are strictly jurisdictional. An FDA approval gives you the green light to market your device in the United States, and that’s it. To sell in the European Union, you need to earn a CE Mark, which means navigating the EU's own tough regulatory frameworks (MDR or IVDR).
While the FDA and EU authorities are both focused on safety and effectiveness, they go about it in very different ways. They have their own submission processes, unique documentation standards, and separate review bodies. Think of them as two different countries with their own distinct languages and customs.
This means you can't just copy-paste your FDA submission for the EU market. A global strategy demands a separate, tailored regulatory plan for every single region you want to enter.
Does Compliance End After Market Launch?
Absolutely not. Getting your device to market is a huge win, but it’s the starting line for ongoing compliance, not the finish line. From the moment your device is cleared, you're responsible for its performance for its entire lifecycle.
This is a serious legal obligation. You have to actively maintain your Quality Management System (QMS) to ensure every device you produce is just as safe and effective as the one that got approved. More importantly, you must conduct rigorous post-market surveillance. This involves actively monitoring your device's performance in the real world, collecting data, and immediately reporting any problems or adverse events to the regulators. This vigilance isn't optional—it's a core part of being a medical device manufacturer.
At PYCAD, we live and breathe medical imaging AI. We help companies integrate sophisticated artificial intelligence to improve diagnostic accuracy and streamline workflows. If you're working on a medical device and need help with anything from data management to model deployment, see how our expertise can accelerate your project.