
Think of medical device post-market surveillance as the ongoing safety check that happens after a device hits the market. It’s a systematic way for manufacturers to keep an eye on how their products perform in the real world, catching potential problems that didn't pop up during the initial, more controlled clinical trials.
This constant monitoring is all about ensuring long-term patient safety and, just as importantly, driving continuous improvements to the device itself.
Why Post-Market Surveillance Matters for Your Device

Pre-market clinical trials are a bit like a new car’s controlled test drive on a closed track. It's an absolutely essential step, but it can't fully predict what will happen on real roads with different drivers, weather, and traffic. The real test begins when the public starts driving it every day.
It's the same story for medical devices. Clinical trials, no matter how thorough, are limited to a specific group of patients in a highly controlled environment. Medical device post-market surveillance is what bridges that gap between the lab and real life. It’s far more than just a regulatory box to check; it’s a critical feedback loop that delivers a steady stream of real-world evidence.
This process is so important for a few key reasons:
- Catching Problems Early: It helps spot rare or long-term adverse events that simply wouldn’t appear in shorter trials.
- Confirming Performance: It verifies that the device works as expected for all kinds of different patients and in various clinical situations.
- Driving Smart Improvements: The data collected informs everything from small tweaks to labeling all the way to major design changes.
Moving Beyond Compliance to Innovation
When done right, a strong PMS system shifts from a reactive chore to a genuine strategic advantage. By methodically gathering and analyzing data from the field, manufacturers get priceless insights into how their devices are actually being used. Understanding the real-world performance and limitations, like the accuracy of consumer-grade medical devices, really underscores why this kind of follow-up is so critical.
This continuous cycle of learning does more than just keep patients safe—it sparks innovation. Real-world data can shine a light on unmet clinical needs or reveal opportunities to enhance the next generation of devices. It ensures products don't just stay safe, but actually get better and more effective over time.
Ultimately, a well-run PMS strategy is about building trust. It shows patients, healthcare professionals, and regulatory bodies that you’re committed to safety and quality long after the product launch, proving your device’s reliability throughout its entire lifecycle.
Understanding the Foundations of PMS

To get a real handle on medical device post market surveillance (PMS), you have to understand what it’s really about. At its core, PMS is the ongoing, systematic process of watching how your device performs after it hits the market. Think of it as a living feedback loop between your company and the real world—a constant conversation that tells you what’s working and what isn’t.
But this process isn’t a one-size-fits-all activity. It’s split into two distinct but deeply connected approaches: proactive surveillance and reactive vigilance. Nailing down this difference is the first real step toward building a PMS system that actually works.
The Two Sides of Surveillance: Proactive and Reactive
Let’s use an analogy. Think about a building's security. Proactive security is the guard making their rounds, checking doors, and watching the camera feeds to stop trouble before it starts. The reactive side is the alarm that screams when a window is smashed, demanding an immediate response to a problem that’s already happened.
Medical device surveillance works in exactly the same way.
- Proactive Surveillance: This is your "guard on patrol." It’s about actively going out and collecting data to confirm your device is still safe and performing as expected. You’re not just sitting back and waiting for a red flag; you’re systematically searching for information that proves everything is okay and looking for subtle trends that might hint at future issues.
- Reactive Vigilance: This is your "alarm system." It kicks in when specific events are reported, like a customer complaint, an adverse event report from a clinic, or a device malfunction. This side of PMS is all about responding quickly to investigate and correct things that have gone wrong.
A truly effective PMS plan blends these two seamlessly. The proactive work gives you a solid baseline understanding of your device’s everyday performance, while reactive vigilance makes sure you can jump on any problems the moment they appear.
Defining Key PMS Activities and Reports
Within this framework of proactive and reactive work, there are specific tasks and documents that make up the nuts and bolts of any PMS plan. You’ll hear these terms all the time in regulatory circles, so knowing what they mean is non-negotiable for compliance.
One of the most critical proactive activities is the Post-Market Clinical Follow-up (PMCF). A PMCF is essentially a continuous effort to keep a device's clinical evaluation up to date. This often means running studies or surveys to gather fresh clinical data on how the device is being used in the wild, confirming it remains safe and effective over its entire lifespan.
All this data, along with other feedback, gets compiled into mandatory reports. The specific report you need to create usually depends on your device’s risk class.
- Post-Market Surveillance Report (PMSR): This is generally for lower-risk devices (like Class I). The PMSR is a straightforward summary of the results and conclusions you’ve drawn from your PMS data analysis.
- Periodic Safety Update Report (PSUR): This is required for higher-risk devices (think Class IIa, IIb, and III). A PSUR is a much heavier lift—a deeply detailed document that provides a full evaluation of all the safety and performance data you've gathered.
Don't mistake these reports for just more paperwork. They are powerful analytical tools.
By systematically organizing and analyzing data in PMSRs and PSURs, manufacturers can transform raw information into a clear picture of their device’s real-world behavior. This structured approach is vital for identifying subtle risks or performance shifts over time.
This entire system creates a vital feedback loop. The insights you gain from proactive PMCF studies and reactive complaint handling flow directly into these reports. From there, the conclusions drive real action—from product improvements to updated labeling—ensuring the device stays safe, effective, and in line with what users and regulators expect. This cycle of monitoring, analyzing, reporting, and improving is what powers effective medical device post market surveillance.
Navigating Global Regulatory Requirements
Getting a medical device through approvals and onto the market feels like crossing a finish line. But in reality, it’s just the start of a marathon. Keeping that device available to patients around the world means navigating a complex and often confusing web of medical device post-market surveillance (PMS) rules.
At the center of this regulatory world are two heavyweights: the U.S. Food and Drug Administration (FDA) and the European Union, with its stringent Medical Device Regulation (MDR). While they both share the ultimate goal of patient safety, how they get there couldn't be more different. For manufacturers, understanding these differences is the key to building a single, effective PMS system that satisfies everyone without creating redundant work.
A Tale of Two Systems: The FDA and EU MDR
The biggest split between the FDA and the EU MDR comes down to a simple question of philosophy: should surveillance be reactive or proactive? The FDA has traditionally leaned towards a data-driven, reactive model, while the EU MDR has fully embraced a proactive, lifecycle-based approach.
The EU's Medical Device Regulation (MDR) 2017/745 truly changed the game, making proactive lifecycle management the core of compliance. This is a stark contrast to the U.S. FDA's more traditional framework, which has focused more on reacting to collected data. If you want to dive deeper, you can get more details on the USA vs EU requirements.
So, what does that look like in the real world?
- The FDA Approach: The U.S. system is built around manufacturers reporting problems after they happen. Think of systems like the Manufacturer and User Facility Device Experience (MAUDE) database. The focus is on spotting trends and signals from real-world data and then acting on them.
- The EU MDR Approach: The EU demands a more forward-thinking process. Manufacturers must constantly and proactively update their clinical evaluation and risk documents throughout the device's entire life. It’s not enough to wait for a problem to pop up; you have to be actively looking for data to prove your device is still safe and performing as expected.
This proactive mandate in the EU results in more structured and frequent reporting, putting the onus on manufacturers to always be collecting and analyzing data.
Comparison of FDA vs EU MDR Post Market Surveillance Requirements
To truly grasp the practical differences, it's helpful to see the requirements side-by-side. The following table breaks down the key distinctions between the FDA and EU MDR frameworks, showing where their approaches to post-market surveillance align and where they diverge.
| Requirement Area | FDA (United States) | EU MDR (European Union) |
|---|---|---|
| Core Philosophy | Primarily reactive, focused on identifying and responding to adverse events and product issues. | Proactive and continuous, requiring ongoing risk-benefit analysis throughout the device lifecycle. |
| Key Reporting | Adverse Event Reporting (MDRs), device tracking for certain high-risk devices, some periodic reports for PMA devices. | Periodic Safety Update Report (PSUR), Post-Market Surveillance Report (PMSR), Post-Market Clinical Follow-up (PMCF) reports. |
| Data Collection | Relies heavily on complaint files, Medical Device Reports (MDRs), and service records. | Mandates a broad range of data sources, including clinical data, user feedback, literature, and proactive PMCF studies. |
| Risk Management | Risk management files are updated primarily when new risks or issues are identified. | Risk management is a "living" process, requiring continuous updates based on all incoming PMS data. |
| Key Documents | Post-Market Surveillance Plan is required, but less prescriptive than the EU's. | Requires a detailed Post-Market Surveillance (PMS) Plan and a Post-Market Clinical Follow-up (PMCF) Plan for most devices. |
| Frequency | Reporting is often event-triggered (e.g., within 30 days of an incident). Periodic reports are typically annual for PMA devices. | Reporting is on a fixed schedule. PSURs are required annually or biennially depending on device risk class. |
This comparison makes it clear that while both regulators prioritize safety, the EU MDR's lifecycle-focused approach demands a more comprehensive and continuously active surveillance system from the outset.
Creating a Harmonized Global PMS Strategy
At first glance, trying to satisfy these two different systems might seem like an impossible task. The secret? Build your global PMS strategy on the most demanding requirements.
If you design your system to meet the proactive, continuous evaluation standards of the EU MDR, you'll almost certainly gather all the data and insights needed to meet the FDA's requirements along the way. Think of it as aiming high—you'll hit all the lower targets automatically.
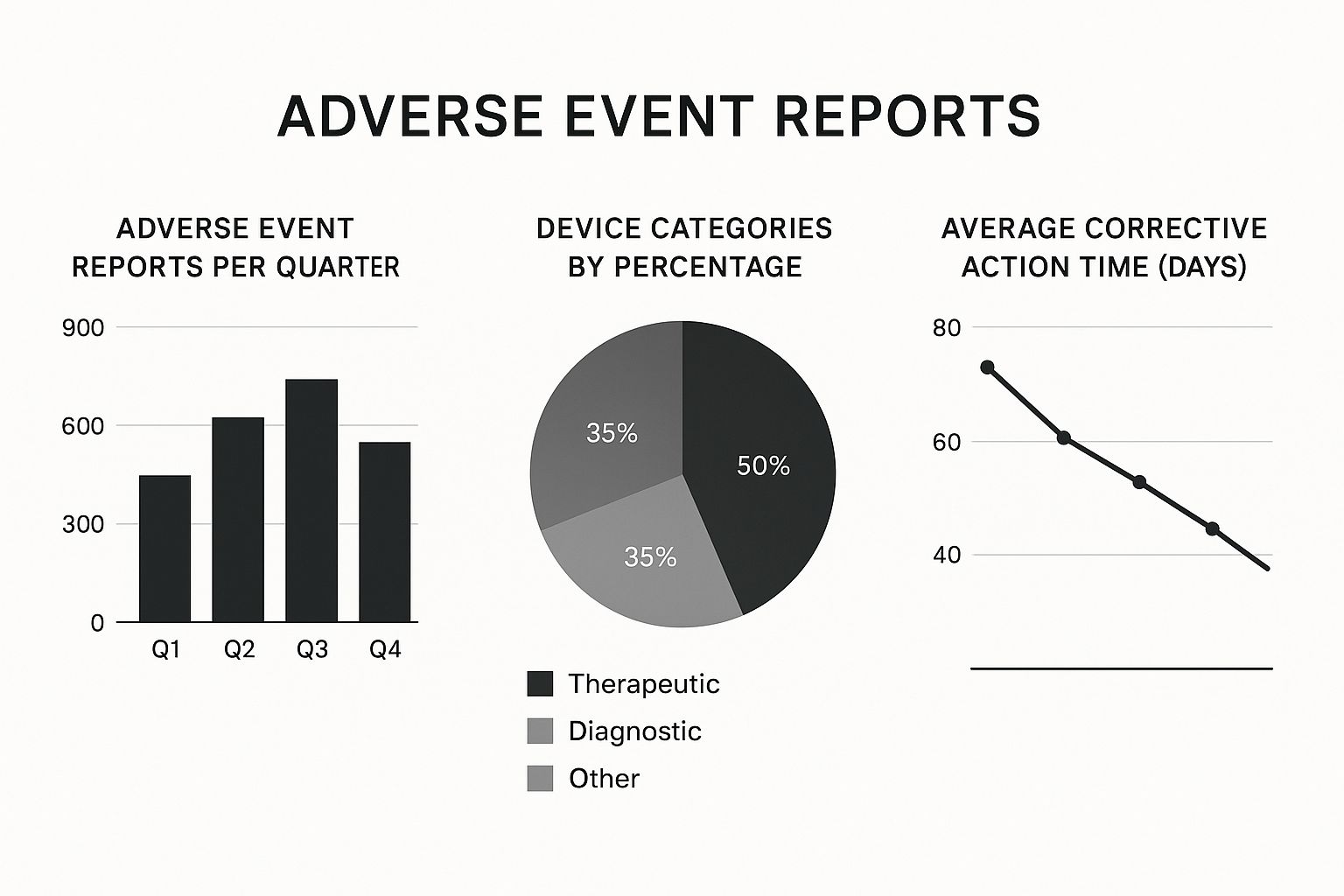
A smart, harmonized approach usually involves these four steps:
- Unify Your Data Collection: Create a single, central system to house all your PMS data. This includes everything from customer complaints and clinical study results to literature reviews, no matter where in the world the data came from.
- Map Out Global Requirements: Create a clear cheat sheet that documents every specific report, deadline, and data point required by every regulatory body you answer to.
- Build Proactive Processes: Make proactive activities the foundation of your system. Post-Market Clinical Follow-up (PMCF) studies, which are mandatory in the EU, are a perfect example. They provide powerful evidence that all regulators value.
- Use a Modular Reporting Structure: Don't reinvent the wheel for every report. Develop templates that can be easily adapted. The detailed analysis you do for an EU PSUR can be repurposed for an FDA submission, saving an incredible amount of time.
By adopting a global mindset from the very beginning, you can turn post-market surveillance from a stressful, fragmented chore into a streamlined and powerful operation. This doesn't just keep you compliant; it gives you a much richer, more complete understanding of how your device is performing for patients worldwide.
How to Collect Real-World PMS Data

Effective medical device post market surveillance lives and dies by the quality of the data you collect. Once your device is out in the wild, information about how it’s really performing comes at you from all directions. The goal is to build a system that can catch all these signals, whether they’re shouted through a formal complaint or whispered in a patient survey.
Think of yourself as a detective on a case. You can't just rely on the official police reports. You need to talk to witnesses, check the security footage, and hunt for the subtle clues others might miss. In the same way, a strong PMS strategy has to blend both passive and active data collection methods to piece together the complete story of your device.
Passive methods are like leaving the door open for information to find its way to you. Active methods, on the other hand, are about you going out and deliberately hunting for specific information.
Passive Data Collection Sources
Passive data collection is the reactive backbone of your surveillance system. This is the information that comes to you without you having to go out and ask for it. These sources are fantastic for spotting immediate problems and are often the first sign that something needs a closer look.
Here are the most common passive sources:
- Customer Complaints and Feedback: This is your front line. Every call to customer support, every frustrated email, every social media mention—it’s all raw data that can point to usability issues, malfunctions, or user confusion.
- Adverse Event Databases: Regulatory bodies like the FDA maintain massive public databases, such as MAUDE (Manufacturer and User Facility Device Experience). Keeping a close eye on these databases for any mention of your device, or even similar ones, is crucial for spotting wider industry trends and potential safety red flags.
- Service and Repair Reports: The data from your own tech support and repair teams is a goldmine. It gives you hard evidence of component failures, unexpected wear and tear, and patterns of malfunction that might never get reported as a formal adverse event.
While this data is easy to get, it has its blind spots. It’s prone to under-reporting—not everyone who has a problem is going to file a formal complaint. It also tends to amplify negative events, which can skew your perception of the device's overall performance. That’s why you have to balance it with active data collection.
Active Data Collection Strategies
Active data collection is all about being proactive. Instead of waiting for data to show up, you design specific activities to answer key questions about your device’s long-term safety and performance. These methods are absolutely essential for meeting the tough requirements of regulations like the EU MDR.
A well-planned active surveillance program is more than just a box-ticking exercise. It's how you systematically prove that your device's benefit-risk profile stays positive over its entire lifespan.
Here are a few powerful active methods you can use:
- Post-Market Clinical Follow-up (PMCF) Studies: These are formal clinical studies you run after your device is already on the market. They are designed to confirm long-term safety and performance, often by zeroing in on specific patient groups or outcomes. For example, a PMCF study for a new hip implant might track patients for 5-10 years to measure the implant's durability and the patient's mobility over time.
- Patient and Physician Surveys: Sometimes, the best way to get an answer is just to ask. Surveys that target users and doctors can give you incredibly specific feedback on things like user satisfaction, real-world usability challenges, and how the device actually fits into a busy clinical workflow—details you’ll almost never find in a complaint file.
- Patient Registries: Registries are organized systems that gather uniform data on a specific patient population, like everyone with a certain heart condition or who has received a particular implant. Tapping into these allows you to collect rich, long-term data on clinical effectiveness and product use, which is invaluable for both future product development and for clinicians making treatment decisions. You can learn more about the role of registries in post-market surveillance.
Active methods give you structured, high-quality data, but they definitely take more time and money to execute. The real magic happens when you build a hybrid PMS system: passive sources act as your early-warning system, while active methods deliver the deep, clinical evidence you need to confirm long-term safety and drive real product improvements.
How to Turn Raw Data Into Actionable Insights
Collecting data for your medical device post-market surveillance is a lot like gathering ingredients for a complex recipe. Piles of raw data—complaints, survey results, clinical reports—are a great start. But those ingredients are pretty useless until you know how to combine and analyze them to create something meaningful.
The real value isn't in having a mountain of information; it's in turning that data into clear, actionable insights that protect patients and drive real-world product improvements. This means moving from simply having data to truly understanding it. It’s about detecting faint signals in the noise, digging deep to find the root cause of an issue, and using modern tools to make sense of it all.
Ultimately, this is how you transform your PMS data from a regulatory chore into a powerful strategic asset.
Finding the Needle in the Haystack with Signal Detection
Signal detection is the art of spotting that "needle in the haystack"—that one piece of data, or a small cluster of events, that points to a potential safety issue. It's a systematic process of monitoring incoming data for patterns that are unusual or just don't fit with what you already know about your device's performance.
Think of it like being a detective reviewing hours of security footage. Most of it is just routine, but you’re looking for that one small, out-of-place event that breaks the pattern. In post-market surveillance, a signal might be:
- A slight uptick in complaints about a specific component failing.
- An unexpected adverse event reported in a new patient population.
- A string of user reports mentioning confusion with a specific step in the instructions.
These signals aren't definitive proof of a problem, but they are critical alerts that tell you exactly where to focus your investigation. Ignoring them is not an option. Each one demands a thorough assessment to determine if it represents a genuine risk.
From Symptom to Solution with Root Cause Analysis
Once you've flagged a credible signal, it’s time to perform a root cause analysis (RCA). This is a structured investigation that moves past the obvious symptoms of a problem to uncover the fundamental, underlying cause.
Just fixing the symptom is like putting a bucket under a leaky pipe—sure, it contains the immediate mess, but it does nothing to stop the leak.
An effective RCA forces you to repeatedly ask "Why?" until you can't go any deeper. Let's say your signal is an increase in device screen failures. The analysis might unfold like this:
- The Problem: The screen is failing. (But why?)
- Immediate Cause: A specific connector keeps coming loose. (Okay, but why?)
- Deeper Cause: The connector housing has a slight manufacturing defect. (Getting warmer… why?)
- Root Cause: The mold used for that housing is wearing out much faster than expected.
By identifying the worn-out mold as the root cause, the solution becomes obvious: replace the mold. This prevents the problem from ever happening again. Just tightening loose connectors would have been a temporary, inefficient, and ultimately ineffective fix.
Root cause analysis is the bridge between identifying an issue and implementing a lasting corrective and preventive action (CAPA). It ensures you’re solving the real problem, not just patching over the symptoms.
The Rise of AI and Machine Learning in Data Analysis
Let's be honest—the sheer volume of PMS data can be completely overwhelming. Manually sifting through thousands of complaint narratives, literature articles, and electronic health records is painstakingly slow and wide open to human error. This is where Artificial Intelligence (AI) and Machine Learning (ML) are becoming essential.
AI-powered systems can chew through enormous datasets in seconds, identifying subtle trends and correlations that a human analyst might easily miss. For example, an ML model can scan thousands of free-text complaint descriptions to flag an emerging issue with a device’s battery life long before it becomes a statistically significant trend in your structured data.
By automating the initial review, these technologies free up your team to focus their expertise where it matters most: investigating the critical signals and turning that raw data into life-saving insights faster than ever before.
Building Your Compliant and Effective PMS System
Putting together a solid post-market surveillance system isn't just about checking regulatory boxes. It’s about building a living, breathing framework that actively protects patients and makes your products better over time. Think of it as a sophisticated listening station, constantly tuned into signals from the real world, interpreting them correctly, and then using that intelligence to make smart, informed decisions.
A truly effective PMS system is so much more than a binder full of documents. It's a deeply integrated process that needs a clear plan, the right people, and seamless collaboration across your entire company.
Your starting point is the PMS plan. This is the blueprint for your entire surveillance strategy. It has to spell out your goals, pinpoint the exact data sources you'll monitor (both active and passive), and lay down clear procedures for how you'll collect, analyze, and report on everything you find.
The Power of Cross-Functional Collaboration
A PMS system just won't work if it's stuck in a silo. Real success happens when you get different departments working together, each bringing their unique expertise to the table. This isn’t just a nice-to-have; it's essential for getting a complete picture of how your device is performing out in the wild.
Picture your PMS system as a central hub with spokes reaching out to key teams:
- Regulatory Affairs: This team keeps the whole operation compliant, making sure the PMS plan meets all global rules and that reports like PSURs are submitted correctly and on schedule.
- Quality Assurance: QA is on the front lines. They’re the ones managing complaints, digging into root cause analyses, and kicking off corrective and preventive actions (CAPAs) when needed.
- Research & Development (R&D): The feedback from PMS data is pure gold for R&D. It helps them spot opportunities for design improvements and shapes the development of next-generation devices.
- Marketing and Sales: These folks are constantly talking to clinicians and end-users. They have an invaluable, on-the-ground perspective on usability and customer satisfaction.
When these teams are in sync, the data you collect becomes far richer and more meaningful. Isolated complaints suddenly transform into a complete story about your device's lifecycle. To get this humming, it's worth exploring the top software for compliance, which can make a huge difference in managing data and regulatory tasks.
Creating a Harmonized Global Strategy
In today’s market, your PMS system has to be multilingual when it comes to regulations. Harmonizing your strategy to meet the standards of different authorities, like the FDA and the EU, is non-negotiable for efficiency and compliance. The trick is to build your system around the strictest requirements—usually the EU MDR—because if you can satisfy them, you're likely covered for the others.
This global mindset is actually getting easier, thanks to international efforts to standardize how we talk about and handle device issues.
Regulatory bodies such as the International Medical Device Regulators Forum (IMDRF) continue to refine their guidance on adverse event reporting, fostering global consistency in categorizing and managing risks. The IMDRF's work on updating adverse event terminology helps simplify worldwide market surveillance and vigilance. You can find out more about these regulatory themes and what they mean for manufacturers by exploring updates on global medical device regulations.
Finally, never forget that meticulous documentation and record-keeping are the foundation of it all. Every procedure, every analysis, and every decision needs to be documented thoroughly. This creates a crystal-clear audit trail that shows you're not just talking about patient safety—you're living it, every single day.
Your PMS Questions, Answered
Even when you feel you’ve got a handle on the basics, putting a medical device post market surveillance (PMS) strategy into practice always brings up a few more questions. Let’s tackle some of the most common ones head-on to clear up any confusion and help you get started.
Think of this as your quick-reference guide. Nailing these core ideas is the first step to building a surveillance system that’s not just compliant, but genuinely effective at protecting patients.
What Is the Real Goal of Medical Device Post-Market Surveillance?
At its heart, the main goal is to keep a constant watch on how a medical device holds up in the real world—long after it leaves the controlled environment of a clinical trial. Those pre-market trials are essential, but they happen with a limited number of patients under ideal conditions. PMS is what happens next.
It’s all about gathering data from a massive, diverse group of actual patients over the entire lifespan of the device. This is how you spot those rare but serious adverse events, uncover performance issues that only show up after years of use, and get the insights needed to make the next version even better. Patient safety is always the endgame.
How Is Post-Market Surveillance Different From Post-Market Clinical Follow-up?
This is a classic point of confusion, but the difference is actually pretty simple.
Picture your entire PMS system as a big library, filled with every scrap of information you can find about your device—complaint files, service reports, published studies, user feedback, everything.
Post-Market Clinical Follow-up (PMCF) is just one specific, very detailed research book on a shelf in that library. It’s a proactive part of your PMS plan where you actively go out and collect new clinical data, often through a formal study, to answer specific questions about long-term safety and performance.
In short, PMCF is a tool you use within your broader PMS framework.
What Are the First Steps to Create a PMS Plan?
Starting a PMS plan can feel overwhelming, but it doesn't have to be. The key is to build it on a solid foundation, one step at a time.
Here’s where to begin:
- Define Your Scope and Goals: First, get crystal clear on what you're trying to achieve for a specific device. What are you monitoring? Your goals will depend on the device's risk class, how it's used, and who the patients are.
- Map Out Your Data Sources: Make a complete list of everywhere you can get information. Split them into two camps: proactive sources (like PMCF studies or user surveys) and reactive sources (like complaint logs or repair data).
- Create Your Procedures: Write down exactly how you'll collect, review, and analyze all this data. Who is responsible for what? What are the timelines? This is your operational playbook.
- Know Your Reporting Deadlines: Your plan must outline exactly what reports you need to create (like PSURs for the EU) and when they are due. Don't let a regulatory deadline sneak up on you.
At PYCAD, we focus on using advanced AI to sharpen medical imaging analysis. This provides the kind of data-driven insights that are crucial for a modern, effective post-market surveillance system. See how our AI solutions can help you better monitor your device's safety and performance.





