
When we talk about medical device risk management, we're talking about a structured, ongoing process. It's about systematically finding, analyzing, and reducing any potential harm a medical device could cause, from the day it's first imagined until it's taken out of service.
Think of it as the safety blueprint. It's the disciplined work that ensures a device's benefits to patients will always, without question, outweigh its potential risks. This isn't just a good idea; it's an absolute must for getting regulatory approval and building a successful product.
Why Medical Device Risk Management Is Non-Negotiable

Risk management is far more than just another compliance checkbox. It’s the very foundation of patient safety and market trust. Consider an architect designing a skyscraper. They don't just hope the foundation is strong; they test every single component to prevent a catastrophic failure. That's precisely the kind of proactive mindset that great medical device risk management brings to the table.
This process is deeply embedded in every stage of a device’s life, starting from the earliest concept sketches and continuing long after launch with post-market feedback. It’s how you demonstrate to patients and regulators that safety isn't an afterthought—it's a core design principle.
A Foundation for Safety and Innovation
At its core, this systematic approach gives you a framework to deal with potential problems before they ever have a chance to cause harm. It fundamentally shifts a company's culture from a reactive "fix it when it breaks" mode to a predictive "prevent it from failing in the first place" strategy.
This structured process is absolutely critical for several reasons:
- Protecting Patient Health: The number one goal is always to minimize the possibility of adverse events and protect patient well-being.
- Achieving Regulatory Compliance: Regulators across the globe, including the FDA and EU authorities, require a robust risk management system just to get in the door.
- Building Stakeholder Confidence: A thoroughly documented risk process shows you’re serious about quality, which builds incredible trust with clinicians, hospitals, and the patients themselves.
The medical device industry is anything but static. As technology improves and demand for better treatments grows, the market is expanding at a staggering rate. Projections show the global medical devices market growing from about USD 678.88 billion in 2025 to over USD 1.14 trillion by 2034. You can dig into these market growth projections on Precedence Research.
While this growth is exciting, it also brings more complex risks, especially with the rise of connected devices and the cybersecurity threats that come with them.
At its heart, risk management answers a fundamental question: "Have we done everything reasonably possible to ensure this device is safe for its intended use?" A well-executed plan provides a confident "yes."
More Than Just a Document
Ultimately, medical device risk management isn’t a one-and-done task that creates a static document. It's a living, breathing process. It demands constant attention and must adapt as new data and real-world feedback come in.
This cycle of analysis, control, and review doesn't stop when the product ships. It continues for the entire time the device is being used, ensuring safety remains the top priority and cementing your company's reputation for quality and reliability.
Understanding the Global Regulatory Playing Field
Bringing a medical device to market isn't just about innovation; it's about navigating a maze of global regulations. It's best to think of these rules not as roadblocks, but as a universal language of safety and quality that regulators across the world expect you to speak fluently. For anyone in this industry, getting a firm grip on the requirements from major players like the U.S. Food and Drug Administration (FDA) and the European Union’s Medical Device Regulation (EU MDR) is non-negotiable.
These frameworks lay down the law for proving your device is both safe and effective. They mandate the creation of a Quality Management System (QMS), which is the operational engine driving everything from initial design and manufacturing to what happens after your device is in the hands of users. A solid QMS isn't just about ticking boxes; it's a strategic tool that embeds quality into your company's DNA.
A Major Step Toward Global Alignment
Something big is happening in the United States. The FDA is moving away from its old Quality System Regulation (QSR) and adopting the new Quality Management System Regulation (QMSR). This is far more than a simple name change—it’s a landmark shift designed to align U.S. standards with the globally recognized benchmark, ISO 13485:2016.
For years, manufacturers selling in both the U.S. and abroad have been stuck juggling two slightly different QMS rulebooks. The new QMSR is a game-changer because it directly incorporates the structure and philosophy of ISO 13485, which most of the world already uses.
This harmonization is a huge relief for global companies. It means you can finally build and maintain a single, powerful QMS that meets the demands of multiple regulators at once, reinforcing the core idea that solid medical device risk management is a universal principle.
This shift simplifies everything. Your risk management files, quality procedures, and documentation become more streamlined, cutting down on redundant work and paving a smoother path to market. It signals a worldwide agreement on what it takes to produce a safe and effective medical device.
Core Philosophies of EU and US Regulations
While the FDA and EU MDR are both laser-focused on patient safety, they don't always take the same path to get there. The EU MDR, for example, is intensely focused on the entire lifecycle of a device. It demands more comprehensive clinical evidence upfront and requires a much more rigorous post-market surveillance plan once the device is in use. It also created the formal role of a "Person Responsible for Regulatory Compliance" (PRRC) to ensure someone is always accountable.
Staying on top of these shifting regulations is a real challenge. A major pain point for many companies is updating their QMS to comply with new rules. In fact, a 2025 industry report revealed that nearly half of all medical device companies felt they weren't ready for the beefed-up QMS requirements of the EU MDR. This really drives home the need for a proactive approach to compliance, a topic covered in detail in Greenlight Guru's industry report.
Despite their differences, both frameworks are built on a shared, unshakable foundation: risk management isn't a one-time task but a continuous process woven into every stage of a device's life.
Key Regulatory Frameworks at a Glance
To help clarify the differences, this table offers a quick comparison of the two dominant regulatory systems in the medical device world.
| Framework Feature | FDA QMSR (United States) | EU MDR (European Union) |
|---|---|---|
| Core Standard | Aligned with ISO 13485:2016, focusing on a risk-based QMS. | Requires a comprehensive QMS compliant with detailed annexes. |
| Risk Focus | Emphasizes risk management throughout the product lifecycle. | Mandates a continuous, lifecycle approach to risk management. |
| Clinical Data | Requires appropriate clinical data to support safety and efficacy. | Demands more extensive clinical evidence and ongoing evaluation reports. |
| Post-Market Role | Requires robust post-market surveillance and complaint handling. | Enforces proactive Post-Market Surveillance (PMS) with detailed reporting. |
Ultimately, whether you're targeting the US, the EU, or both, your success hinges on understanding these nuances and building a risk management system that is robust, integrated, and always ready for scrutiny.
Applying the ISO 14971 Risk Management Process
When you're building a medical device, simply wanting it to be safe isn't enough. You need a structured, repeatable, and globally recognized framework to get you there. That's precisely the role of ISO 14971—it’s the definitive international standard for medical device risk management.
Following this standard isn't about getting bogged down in bureaucracy. Think of it as a logical, practical workflow that turns abstract safety goals into a concrete plan of action. It guides you from the first napkin sketch all the way through to post-market surveillance, ensuring safety is a core component of every single decision.
Stage 1: The Risk Analysis Phase
Everything kicks off with risk analysis. This is the detective work, where your team systematically identifies every conceivable thing that could go wrong with your device. You have to consider its entire lifecycle, from manufacturing to disposal.
For any modern connected device, a crucial piece of this puzzle involves mastering security risk assessment. After all, a software vulnerability can be just as dangerous as a physical defect.
Let's imagine we're developing an AI tool that analyzes chest X-rays. What could go wrong?
- Software Bugs: The algorithm might flag a healthy patient with a disease (a false positive), causing unnecessary stress and follow-up procedures.
- Algorithmic Bias: The AI could be less accurate for certain demographics if the training data wasn't diverse enough.
- Cybersecurity Flaws: A hacker could gain access to sensitive patient imaging data, leading to a major privacy breach.
Once you’ve identified a potential hazard, the next job is to estimate the associated risk. This comes down to two key factors: the severity of the harm if it happens, and the probability of it actually happening.
This simple chart illustrates the basic flow of analyzing and evaluating these risks.
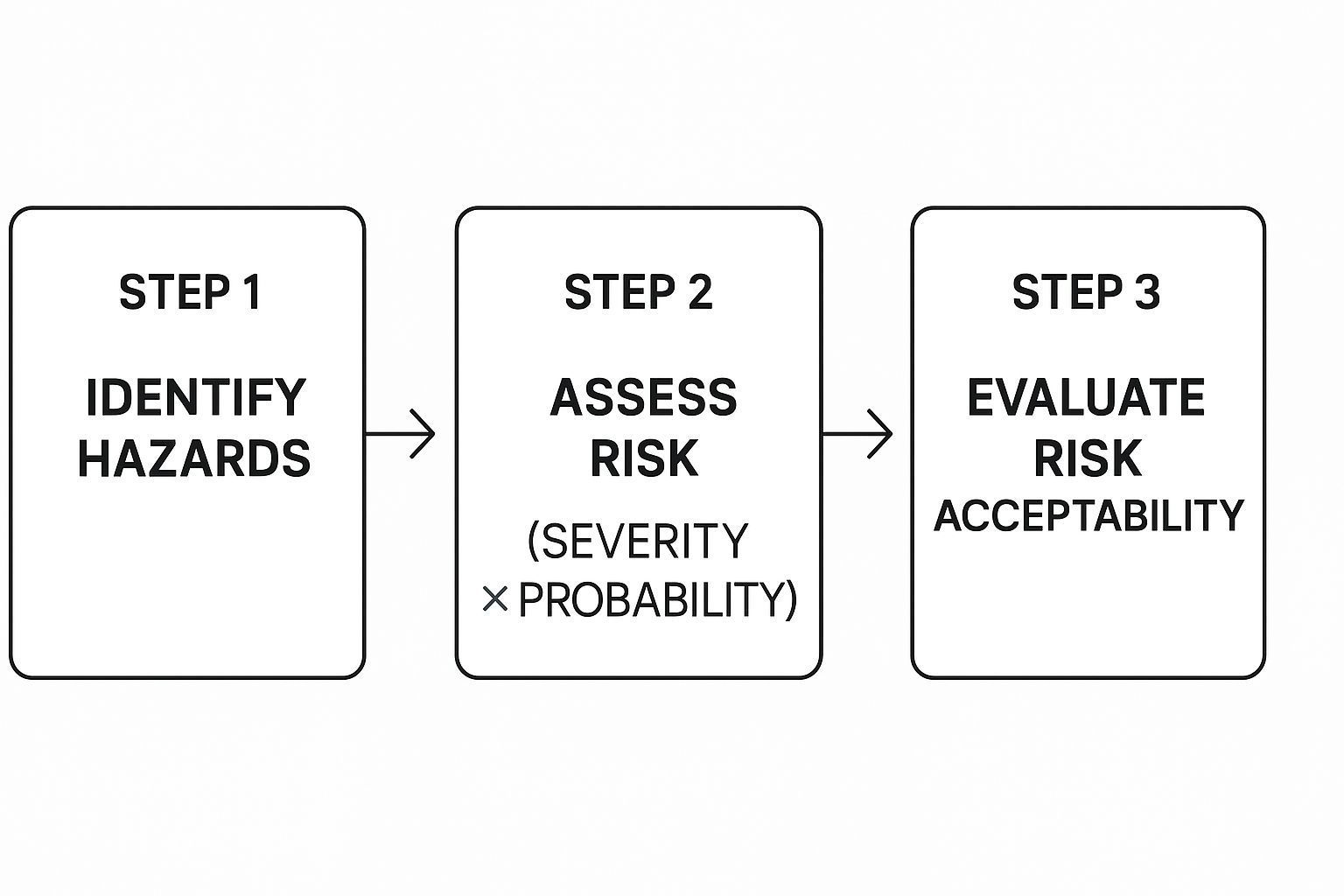
As you can see, just spotting a hazard is the beginning. The real work is in carefully weighing its likelihood and potential impact to decide if it's a risk worth taking.
Stage 2: The Risk Evaluation and Control Phase
With your list of analyzed risks in hand, you move into risk evaluation. This is where you hold each risk up against the acceptability criteria you defined in your Risk Management Plan. Essentially, you ask a simple question: Is this risk acceptable?
If the answer is no, it's time to implement risk controls. This is the engineering phase, where you actively design solutions to bring that risk down to an acceptable level.
A core tenet here is reducing risk to be "as low as reasonably practicable" (ALARP). This acknowledges that zero risk is a myth. The goal is to do everything feasible to minimize it.
Going back to our AI diagnostic tool, here’s how we might control those risks:
- For the Software Bug: We could implement a "human-in-the-loop" workflow, requiring a qualified radiologist to confirm any positive finding from the AI before a diagnosis is finalized. This adds a crucial safety net.
- For Algorithmic Bias: We'd need to proactively source more diverse training data and then run specific validation tests to confirm the model performs equitably across different patient groups.
- For the Cybersecurity Flaw: Strong countermeasures are key. This means enforcing multi-factor authentication, encrypting all patient data (both in transit and at rest), and hiring ethical hackers for regular penetration testing.
Stage 3: Assessing Residual Risk and Benefit
No risk control is perfect. Even after you’ve put your safety measures in place, there will almost always be some small amount of risk left over. This is called residual risk.
The final, critical step is to evaluate this leftover risk. You have to meticulously document and analyze it to see if it now falls within your acceptable limits. If not, it's back to the drawing board to find better or additional controls.
If a residual risk still exists but simply can't be designed out any further, you must perform a formal risk/benefit analysis. This is where you make a compelling, evidence-based case that the device's medical benefit to the patient massively outweighs the total remaining risk. For our AI tool, the benefit of faster, more accurate diagnoses might outweigh the tiny residual risk of a system error—especially with an expert human providing the final sign-off. This documented analysis is a cornerstone of any regulatory submission. It proves you've made a conscious, well-reasoned decision about your device's safety.
Putting Practical Risk Control Measures into Action

Once you’ve pinpointed and evaluated a potential risk, you can't just stop there. The next move in medical device risk management is to actively control it. This isn't a random process; it's a deliberate and structured approach that follows a clear hierarchy. The ultimate aim is to knock that risk down "as low as reasonably practicable" (ALARP), and the only way to do that effectively is to start with your most powerful options first.
The most potent and respected risk control measure is inherent safety by design. It's the gold standard. This means you eliminate the hazard at its source, literally designing it out of existence so the harm can't happen in the first place. Think of it this way: it’s better to build a bridge with sturdy guardrails from day one than to simply put up a sign warning people not to fall off.
For instance, if a device has a sharp edge that could cut a clinician, the best fix isn't a warning sticker. It’s to go back to the drawing board and redesign the casing to be smooth and rounded. This method is incredibly effective because safety is baked right into the device's DNA—it doesn't depend on someone remembering to be careful.
Adding Protective Layers
Of course, designing away every single risk is often impossible. When a hazard is unavoidable, you move to the next level of the hierarchy: adding protective measures. These are the safeguards, fail-safes, and alarms that stand between the hazard and the person it might harm.
Let’s look at an infusion pump. You can't completely design away the fundamental risk of an incorrect dose, but you can build in multiple layers of protection. These could include things like:
- Physical Guards: A simple cover that stops someone from accidentally pressing a critical button.
- Software Limits: A built-in function that prevents a user from programming a dose that is dangerously high or low.
- Alarms: Loud beeps and flashing lights that go off if the pump malfunctions or the IV line gets blocked.
These controls don't make the hazard disappear, but they create active safety nets that dramatically lower the chances of something going wrong. They are the essential second line of defense in any solid risk management plan.
The Last Resort: Information for Safety
The final tier of control is providing information for safety. This covers everything from warning labels and detailed instructions for use (IFU) to user training programs. While absolutely necessary, these are considered the least effective measures because their success hinges entirely on the user. They have to read, understand, remember, and correctly follow the instructions every single time.
A warning label is often a last-ditch effort. Relying on one as your primary control means you're basically shifting the responsibility for safety from your shoulders to the user's, a practice that regulators are never happy to see.
This isn't to say safety information is unimportant—it's critical. A well-written manual can prevent foreseeable misuse. But it should always be the final layer of protection you add, after you've exhausted all design and protective options. A label can clarify a residual risk, but it can never be the solution for a serious one.
Hierarchy of Risk Control Measures
To help visualize this, here’s how the control measures stack up. A robust risk management process always prioritizes the options at the top of the list, only moving down when a higher-level control isn't feasible.
| Control Level | Description | Example |
|---|---|---|
| Inherent Safety by Design | Eliminating the hazard at the source by changing the device's fundamental design. | Using biocompatible materials that cannot cause an allergic reaction. |
| Protective Measures | Adding safeguards, alarms, or fail-safes that protect against the hazard. | An automated shutdown feature on a surgical laser if it overheats. |
| Information for Safety | Providing warnings, instructions, and training to inform users of residual risks. | A label on a device warning against its use near strong magnetic fields. |
After implementing a control—any control—the job isn't done. You must verify that it actually works as intended. Did the design change solve the problem? Does the alarm sound when it’s supposed to? This verification step is non-negotiable; it’s how you close the loop and prove you've successfully brought the risk down to an acceptable level.
Managing AI and Cybersecurity Risks in Modern Devices
Medical devices aren't just standalone tools anymore. They've become intelligent, connected systems that constantly communicate, forming what we now call the Internet of Medical Things (IoMT). While this evolution brings incredible advantages for patient care, it also throws open the door to a whole new world of risks—risks that traditional risk management frameworks were never designed to address.
This shift forces us to look beyond just physical or mechanical failures. The new frontier is all about software vulnerabilities, data integrity, and the very logic of artificial intelligence. A "threat" isn't just a sharp edge or a faulty component anymore; it can be a single line of malicious code or a weakness in a hospital's network firewall.
The New Face of Device Risk
The risks in today's connected devices are a different breed entirely. They're often invisible and can ripple out to affect not just one patient, but entire healthcare networks. A robust risk management process has to grow to include these digital-age hazards.
Two of the biggest new risk categories you need to have on your radar are:
- Algorithmic Bias: Imagine an AI diagnostic tool trained on a dataset that isn't diverse. It might perform poorly for certain patient groups, leading to misdiagnoses and deepening health inequities. This is a real and growing concern.
- Cybersecurity Breaches: A patient monitor with weak security is a prime target. A successful hack could expose massive amounts of protected health information (PHI) or, in a nightmare scenario, allow an attacker to tamper with the device's function.
These technologies are fundamentally reshaping the industry. We're seeing a huge push toward AI-powered robotic surgery and predictive diagnostics. But as the IoMT connects more devices, it also dramatically widens the attack surface for cyber threats. You can get a deeper dive into these advanced medical device industry dynamics on Galendata.com.
Integrating Cybersecurity into Your Risk File
Regulators like the FDA are sending a crystal-clear message: cybersecurity isn't an afterthought. It has to be a central part of your risk management activities from the very beginning. You can't just pass security off to the IT department anymore—it's a critical patient safety issue.
What does this mean in practice? It means you have to build threat modeling and vulnerability assessments directly into your risk management file, right alongside your traditional hazard analysis. Think of it this way: you already anticipate how a physical part might wear down and fail. You need to apply that same foresight to how a hacker might try to break in.
When developing a connected device, you must assume it will face a cyber threat. This proactive stance forces you to build in security from the beginning, rather than trying to patch it on later.
To get ahead of these risks, organizations need to implement comprehensive security measures. This includes not just technical safeguards for the device itself but also broader initiatives like specialized security awareness training for healthcare professionals, which addresses the unique challenges of the medical field.
At the end of the day, protecting a modern device means protecting its physical integrity and the data it handles. In today's world, a successful risk strategy must be just as focused on stopping a data breach as it is on preventing a mechanical failure.
Building a Living Risk File with Post-Market Surveillance

Getting a medical device to market feels like crossing the finish line, but in reality, it's just the start of the race. Your Risk Management File isn't a trophy to be put on a shelf; it's a living, breathing document that has to evolve with every new piece of information you gather once your device is out in the world.
This is where Post-Market Surveillance (PMS) comes in. Think of it as your intelligence network in the field. PMS is the formal, structured process for collecting and making sense of real-world experiences with your device. It’s what turns passive waiting into proactive risk management and is absolutely essential for keeping a device safe throughout its entire lifecycle.
The Real-World Feedback Loop
Let’s be honest: your initial risk assessments are educated guesses. They're based on controlled tests, simulations, and what you predict will happen. Post-Market Surveillance, on the other hand, is driven by what actually happens. This real-world data is infinitely more valuable.
This critical information flows in from multiple channels, each giving you a different angle on your device's performance and safety. This incoming data is the very lifeblood of your living risk file, providing the triggers you need to go back and challenge your original assumptions.
Key data sources include:
- User Complaints and Feedback: Direct-from-the-source reports from clinicians and patients about anything from confusing instructions to unexpected side effects.
- Service and Repair Reports: Maintenance logs are a goldmine. They can reveal patterns of component failure or wear that you never could have anticipated in a lab.
- Published Clinical Studies: New academic research on your device—or even on similar ones—can uncover long-term effects or risks you didn't know existed.
- Vigilance Reporting: These are the mandatory reports you send to regulators about serious incidents. They are critical, high-signal data points.
Each report, each call, each study helps you build a much sharper, more accurate picture of how your device behaves in the messy, unpredictable real world.
A single complaint might seem like an outlier. But when you spot five more just like it, you’re not looking at an anomaly anymore—you’re looking at a trend. Your PMS system needs to be sharp enough to detect these early signals before they become major problems.
From New Data to Action
When this new information arrives, the medical device risk management cycle starts all over again. The data isn't just filed away; it has to be formally reviewed to see how it affects your existing risk analysis. Does a new complaint point to a hazard you completely missed? Does a spike in repair requests mean a known failure is happening more often than you calculated?
If the answer is yes, your Risk Management File must be updated. And this is far more than a paperwork drill—it leads to real-world changes. A string of complaints about a confusing user interface might force a redesign of the software workflow or the creation of better training materials.
This constant loop of surveillance, review, and action is how you ensure your risk controls are still working and your risk/benefit balance remains favorable. It's how you demonstrate, year after year, that your device is safe and effective, maintaining the trust you've worked so hard to build with patients and regulators alike.
Frequently Asked Questions
As you get your hands dirty with medical device risk management, a few key questions almost always pop up. Let's walk through some of the most common ones to clear up any confusion and help you navigate the practical side of things.
What Is the Difference Between a Hazard and a Risk?
It's easy to use these terms interchangeably, but in the world of medical devices, they mean very different things.
Think of a hazard as a potential source of harm. It's a static condition. For example, a sharp edge on a surgical tool, a software bug, or a biocompatibility issue with a material are all hazards. They just sit there, waiting for something to happen.
Risk, on the other hand, is where the action is. It’s the probability of that hazard actually causing harm, combined with the severity of that harm. The sharp edge (the hazard) might not be a big deal if it's tucked away inside the device. But if it’s on the handle where a surgeon's hand will be, the chance of a severe cut makes the risk skyrocket. We don't manage hazards; we manage risks.
How Often Should We Update Our Risk Management File?
Your Risk Management File isn't a "set it and forget it" document. It’s a living file that has to evolve right alongside your device. You must update it anytime new information changes your understanding of the device's risk profile.
While most teams schedule a formal review at least once a year, you can't wait that long if something significant happens. A major event should trigger an immediate update to ensure the file always reflects the real-world safety of your device.
So, what counts as a trigger?
- Design or Manufacturing Changes: Any tweak to the device, no matter how small you think it is.
- Post-Market Feedback: This is huge. User complaints, service reports, or new clinical data are goldmines of risk information.
- New Regulations: When the goalposts move, you have to adjust your game plan.
- Evolving "State of the Art": If a new technology or safety standard becomes best practice in the industry, you need to assess how it impacts your device.
Is It Possible to Eliminate All Risks?
In a word, no. It's completely impossible to get rid of every single risk tied to a medical device.
The real goal of risk management is to reduce every identified risk to an acceptable level. This is guided by a core principle you'll hear a lot: "as low as reasonably practicable" (ALARP). You have to do everything you reasonably can to minimize risk, but you can’t achieve zero.
The critical next step is to formally prove that the medical benefits your device offers patients clearly outweigh the total residual risks that remain. This risk/benefit analysis is the heart and soul of your regulatory submission. It shows regulators you’ve made a conscious, well-documented decision about your device's safety.
What Is the Role of a Risk Management Plan?
The Risk Management Plan is your master blueprint. Mandated by ISO 14971, this document lays the foundation for every single risk-related activity for your device.
You have to create this plan right at the start of a project. It defines the scope of your work, spells out who on the team is responsible for what, and—most importantly—sets the specific criteria for risk acceptability. It's the roadmap that details how you'll verify your controls are working and how you'll collect post-market data, keeping everyone aligned from day one.
At PYCAD, we live and breathe the challenges of integrating AI into medical imaging. We help teams enhance diagnostic accuracy while upholding the strictest standards of safety and risk management. With our deep expertise in data handling and model deployment, we can ensure your innovative devices are built on a solid foundation of quality. Find out how we can help with your next project by visiting us at https://pycad.co.





