
Developing software for a medical device isn't like building your typical app. It's a highly disciplined process of designing, coding, testing, and maintaining software that is either embedded within a physical device or acts as a medical device all on its own. Everything you do must be guided by a strict adherence to regulatory standards, such as IEC 62304 and FDA guidelines, because patient safety and clinical effectiveness are the ultimate goals.
This field is where meticulous software engineering meets even more rigorous risk management and quality control.
The New Era of Medical Device Software Development

Code is fundamentally changing how healthcare works. It's moved far beyond just handling administrative tasks and now plays a direct role in diagnosis, treatment, and ongoing patient monitoring. This shift is being pushed by powerful market demands, especially the rise of personalized medicine and the clear need for reliable remote care.
For developers, this means you have to think differently. The process of creating software in this space is worlds away from standard application development.
SaMD vs. SiMD: The Two Faces of Medical Software
To get your bearings in this field, you first need to understand the two main categories of medical software. They might sound similar, but they are worlds apart in practice.
- Software as a Medical Device (SaMD): This is software that functions as a medical device entirely on its own. It's not part of any hardware. A great example is a mobile app that analyzes a picture of a mole to check for signs of melanoma.
- Software in a Medical Device (SiMD): This is the embedded software or firmware that makes a physical medical device work. The code running an MRI machine or the logic inside an insulin pump are classic examples of SiMD.
Getting this distinction right is critical because it determines your entire development process and regulatory strategy. A mobile diagnostic app and an implantable device have completely different risk profiles, and you can't approach them the same way.
The difference between SaMD and SiMD dictates everything from your initial design controls to your final regulatory submission. Understanding which category your product falls into is one of the first and most important decisions you'll make. Here’s a quick breakdown to help clarify the key distinctions.
SaMD vs SiMD Key Differences
| Characteristic | Software as a Medical Device (SaMD) | Software in a Medical Device (SiMD) |
|---|---|---|
| Primary Function | Performs a medical function independently of any hardware device. | Controls or supports the function of a hardware medical device. |
| Platform | Runs on general-purpose computing platforms (e.g., smartphones, servers, PCs). | Embedded within the specific hardware it controls. |
| Typical Examples | – AI-powered image analysis for X-rays – Mobile app for diabetes management – Clinical decision support software |
– Firmware for an infusion pump – Software running a pacemaker – Code controlling a CT scanner |
| Regulatory Focus | On the software's performance, safety, and clinical claims. | On the entire system (hardware + software) and their interaction. |
| Development Cycle | Can be more agile and iterative, similar to other software products. | Often tied to the hardware development lifecycle, which can be longer. |
Ultimately, whether you're building SaMD or SiMD, the core principle is the same: if the software performs a medical function, it is a medical device and must be developed with that level of rigor. There are no shortcuts.
The core principle is simple: if the software performs a medical function, it must be developed with the rigor of a medical device. There are no shortcuts when patient safety is on the line.
Market Growth and The Push for Innovation
The global market for SaMD alone tells a powerful story. In 2025, it's expected to hit around $36.92 billion, but projections show it rocketing to $210.74 billion by 2035. That's a compound annual growth rate of nearly 19%, driven largely by breakthroughs in AI and the move toward cloud-based healthcare. You can dive deeper into this data by reviewing the full market analysis from Roots Analysis.
This growth is also being fueled by a serious push from government and research bodies to speed things up. Take, for example, ARPA-H’s initiative to revolutionize clinical trials and medical R&D. Programs like this are designed to shrink the timeline from a great idea to a real-world patient solution. This puts even more pressure on development teams to be both efficient and flawlessly compliant from the very first line of code.
Navigating Regulatory Compliance and Design Controls
When you're building medical software, the old "move fast and break things" mantra goes right out the window. Instead, your world revolves around a framework where every single decision is deliberately made, documented, and justified. This isn't about creating red tape for the sake of it; it's about building a disciplined process that puts patient safety and product effectiveness at the very center of everything you do.
You'll quickly become familiar with two critical frameworks: the FDA's Design Controls under 21 CFR 820.30 and the international standard for software lifecycle processes, IEC 62304. Don't think of these as a final checklist. Treat them as the foundational blueprint for your entire project.
The Heart of Design Controls and IEC 62304
If there's one word that captures the essence of both the FDA and IEC 62304 guidance, it's traceability. You absolutely must be able to draw a clear, unbroken line from an initial user need all the way down to a specific line of code and the test that verifies it. This creates a powerful chain of evidence showing your software does exactly what you claim it does, and safely.
Let’s imagine your team is developing software for a new diagnostic imaging tool. A radiologist gives you a critical user need: "I must be able to measure a tumor with an accuracy of +/- 0.5mm." This single sentence sets off a cascade of required design control activities.
Your team will take that need and translate it into formal software requirements, then into architectural designs, and finally, into code. Every step of this journey gets meticulously documented in what’s called the Design History File (DHF). This file becomes the official, living record of your development process, and it’s exactly what an auditor will comb through to confirm you followed a controlled, compliant process.
This infographic shows how fundamental activities like defining requirements and managing risk are prerequisites to even thinking about a regulatory submission.
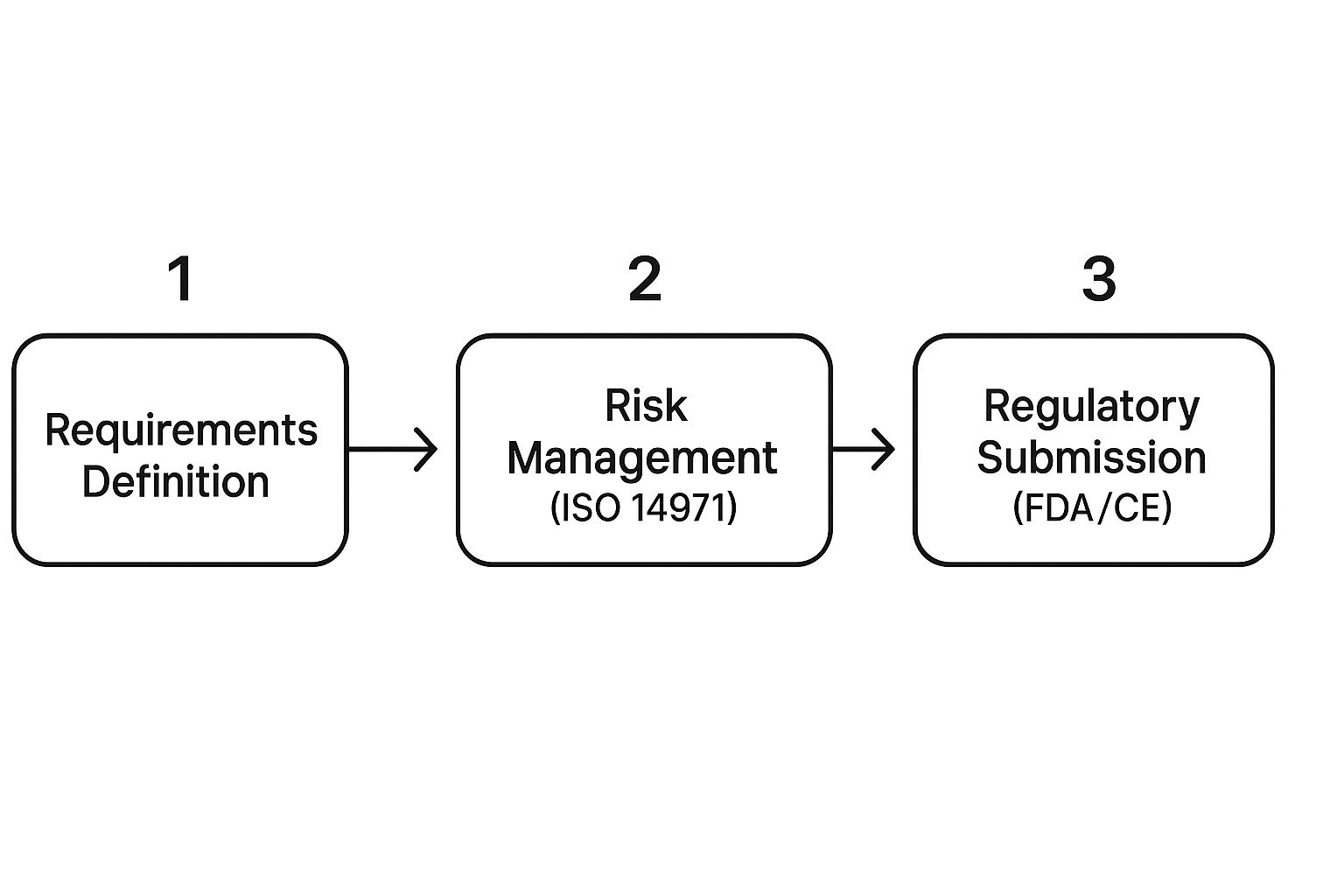
As you can see, risk management isn't a side task. It's woven directly into the process of understanding and defining what you're building in the first place.
Understanding Software Safety Classifications
A huge part of complying with IEC 62304 is figuring out your software's safety classification. This isn't just an academic exercise—it directly dictates the level of rigor and documentation your project demands. The classification hinges on one simple question: what is the worst possible harm that could result if the software fails?
There are three classes:
- Class A: No Injury or Damage to Health is Possible. The software can't contribute to a hazardous situation. Think of an app that just displays patient educational pamphlets.
- Class B: Non-Serious Injury is Possible. A software failure could lead to a hazardous situation resulting in minor harm. Software that analyzes blood glucose readings to suggest lifestyle changes might land here.
- Class C: Death or Serious Injury is Possible. A failure here could be catastrophic. The software controlling a radiation therapy machine or our diagnostic imaging tool from earlier would absolutely be Class C.
Getting this classification right is one of the most important things you'll do early on. If you over-classify, you'll sink time and money into unnecessary documentation. But if you under-classify, you've created a massive compliance gap, put patients at risk, and all but guaranteed a rejection from regulators. For our imaging tool, its central role in planning treatment firmly plants it in Class C, requiring the highest possible level of documentation and testing.
The point of design controls isn't to create more paperwork. It's to build a 'living' record of your design choices. This Design History File (DHF) is your proof that you developed the device in a controlled and systematic way—one that can stand up to any audit.
Building a Compliant Design History File
Think of the DHF as the complete story of your product, from the first sketch on a napkin to the final released version. It's the collection of every document your team generates throughout the software's lifecycle. It has to be organized, easy to navigate, and absolutely complete.
For our imaging software project, a solid DHF would contain things like:
- User Needs and Design Inputs: The original requirements gathered from radiologists, clinicians, and even patients.
- Design Outputs: All the documents that describe the software itself, such as architecture diagrams, commented code, and interface specifications.
- Design Verification and Validation: The full suite of test plans, test cases, and the objective evidence proving the software does what you said it would.
- Design Reviews: Official records from formal meetings where the team reviewed the design at key milestones to make sure it was on track.
- Design Transfer Documents: The "recipe" for installing and deploying the validated software correctly onto the final hardware.
Successfully getting through this regulatory maze means baking these documentation habits directly into your development workflow. When compliance becomes a natural byproduct of a high-quality process, you not only satisfy the regulators but you also end up with a much better, safer, and more effective medical device.
Building a Bulletproof Risk Management Strategy

When you're building medical software, risk isn't just a business concern—it's a direct threat to patient safety. This is why a proactive, highly structured approach to risk management isn't just a good idea; it's the absolute foundation of any compliant and successful product. Your playbook for this entire process is ISO 14971, the global standard for applying risk management to medical devices.
This goes far beyond simply making a list of what could go wrong. It’s a rigorous, systematic process. You have to identify potential harms, figure out how likely they are to happen and how severe the consequences would be, and then implement solid controls to bring those risks down to an acceptable level. Getting this wrong can lead to devastating product recalls, stiff regulatory penalties, and, worst of all, patient harm.
Putting ISO 14971 into Practice for Software
The heart and soul of ISO 14971 is the Risk Management File (RMF). Don't think of this as a single document. It’s a living, breathing collection of records that chronicles every risk-related activity for your device. This file is your proof—the objective evidence that you have a continuous process for finding, evaluating, and controlling risks throughout the product’s entire life.
A truly effective risk analysis demands that you think on two distinct levels:
- System-Level Hazards: These are the big-picture sources of harm tied to the device's overall function and how it's used. For instance, a hazard for a diagnostic imaging system might be the "misinterpretation of an image leading to an incorrect diagnosis."
- Software Failure Modes: This is where you get granular. These are the specific ways the software itself can malfunction and contribute to that system-level hazard. For that same imaging system, a software failure mode could be an "algorithm incorrectly calculating the size of a detected anomaly."
Making this distinction is absolutely critical. You have to be able to draw a clear line from a specific software bug all the way to a system-level hazard and the potential harm it could cause a patient.
The Ever-Growing Threat of Cybersecurity
In the past, risk management was mostly about functional failures—what happens if the software just doesn't work right? Today, that's only half the battle. Cybersecurity has shifted from being an IT problem to a frontline patient safety issue. The FDA has been crystal clear in both its premarket and postmarket guidance: strong cybersecurity controls are a mandatory part of medical device software development.
A malicious attack on a medical device can be catastrophic, leading to anything from a breach of sensitive patient data to the complete shutdown of a life-sustaining device. It's no surprise that a recent study found the healthcare industry was the most targeted sector for cyberattacks in 2022, which really highlights how urgent this is.
A secure software development lifecycle (sSDLC) isn't a 'nice-to-have' anymore. It's a non-negotiable requirement. Regulators expect to see evidence that you've designed security into your device from the very first line of code, not just bolted it on at the end.
Creating a Secure Software Development Lifecycle
Folding security into your development process isn't a one-and-done task. It's a continuous effort that has to be woven into every single phase of the product lifecycle.
Here are some key activities that need to become second nature:
- Threat Modeling: Very early in the design phase, get the team together and brainstorm potential threats. Who are the potential attackers? What would they want? Where are the weakest points in your system? This helps you design security in, proactively.
- Vulnerability Management: You need a process for constantly finding and fixing vulnerabilities, both in your own code and in any third-party components you use. This means running static and dynamic code analysis and regular software composition analysis (SCA).
- Secure Coding Practices: Your developers need to be trained on secure coding standards to avoid common pitfalls like buffer overflows or weak authentication.
- Penetration Testing: Before you go to market, bring in the pros. Hire ethical hackers to hammer on your device's defenses and find the holes your internal team might have missed.
The stakes are only getting higher as this market explodes. The entire medical device industry, which is now deeply intertwined with software, is seeing massive growth. The global medical devices market was valued at an incredible USD 678.88 billion in 2025 and is set to grow at a CAGR of 6%. This boom is driven by new technology and a soaring demand for better diagnostic and monitoring tools—most of which rely on complex software. You can dive deeper into these trends by checking out the latest medical device industry statistics.
This growth means more devices, more connectivity, and a much larger attack surface for bad actors. Your risk management file must now thoroughly document cybersecurity risks, your analysis of them, and how effective your controls are, with the same rigor you apply to any other potential harm. A truly bulletproof strategy is one that marries classic safety risk analysis with modern cybersecurity diligence.
Mastering the Software Verification and Validation Process
How do you actually prove your software works as intended? It’s one thing to build it, but it’s another entirely to demonstrate that you’ve built it correctly and that it’s the right product for its clinical purpose. This is the core challenge of software verification and validation (V&V), the phase where you generate the hard, objective evidence regulators need to see.
Though people often lump them together, verification and validation are two distinct concepts. Confusing them is a common mistake, but getting them right is non-negotiable for a smooth regulatory submission.
-
Verification: This asks, "Did we build the software right?" Essentially, you're checking your work against your own blueprints—the software requirements and design specifications. It's an internal-facing process to confirm the code does what you designed it to do.
-
Validation: This asks a much bigger question: "Did we build the right software?" Here, you're confirming that the final product actually meets the user's needs and intended use in a real-world (or simulated real-world) clinical setting. This is about making sure you solved the correct problem for the user.
One of the biggest pitfalls I see teams fall into is treating V&V like a final exam you cram for at the end. That approach is a recipe for disaster. Instead, think of it as a continuous process woven into the entire development lifecycle. Catching issues early saves a tremendous amount of time, reduces costly rework, and builds a rock-solid case for your device's safety and effectiveness from day one.
Creating a Comprehensive V&V Plan
Your V&V plan is your strategic playbook for all testing. It’s the document that lays out the scope, methods, and acceptance criteria for both verification and validation. More importantly, it ensures every single test has a clear purpose and traces directly back to a specific requirement. When an auditor comes knocking, they’ll want to see this plan and the meticulously documented evidence that you followed it.
A good plan breaks down the different layers of testing you'll perform. Each layer builds on the last, adding another level of confidence in the software's integrity.
-
Unit Testing: This is the ground floor. Developers test individual functions, methods, or classes in isolation to make sure each tiny piece of code behaves exactly as it should. It's the first line of defense against bugs.
-
Integration Testing: Once the individual units are verified, you start combining them and testing how they interact. This is where you uncover issues at the seams—the interfaces and data handoffs between different software components.
-
System Testing: Now we’re looking at the whole picture. System testing involves evaluating the complete, integrated software to confirm it meets all its specified requirements as a cohesive whole, usually in a simulated end-user environment.
From Test Cases to Traceability
Strong test cases are the lifeblood of your V&V effort. A well-written test case is clear, repeatable, and has a pass/fail criteria that leaves no room for ambiguity. Crucially, every single software requirement must be covered by at least one test case that proves it has been met. This creates an unbroken chain of evidence from requirement to test result, known as a traceability matrix.
Let's imagine a requirement for a blood glucose monitor: "The software shall display a visual alert if the glucose reading is above 180 mg/dL." A solid test case would methodically check inputs like 179, 180, and 181 to prove the alert triggers precisely when it should, and not when it shouldn't.
The entire point of V&V is to generate objective evidence. This isn't about feelings or developer confidence; it's about producing documented, repeatable proof that your software is safe, effective, and compliant.
This is where automated testing becomes an indispensable partner. While you'll always need some manual testing (especially for things like user experience), automation can dramatically boost your efficiency and coverage. Automated scripts can run thousands of tests in hours, giving you fast feedback and flagging any regressions introduced by new code.
For high-risk Class C devices, where a software failure could lead to serious injury or death, a robust suite of automated tests isn't a nice-to-have—it's an absolute necessity. Your V&V plan must clearly state which tests will be automated and which will remain manual, along with a solid justification for that approach.
Weaving AI and Machine Learning into Your Medical Device

Artificial intelligence isn’t some far-off concept anymore. It's here, and it’s fundamentally changing what medical technology can do. Bringing AI and machine learning (ML) into a medical device can unlock incredible potential, from spotting diseases before they're obvious to creating therapies that adapt in real-time.
But let's be clear: this adds a whole new layer of complexity to an already tough development process. You're not just writing code. You're building dynamic, learning systems that have to be provably safe and effective inside a rigid regulatory framework. This is the central challenge—how do you validate something that's designed to change?
A New Regulatory Playbook for Learning Algorithms
Regulators like the FDA get it. They understand that AI-driven software is a different animal. A static algorithm is one thing, but an ML model that evolves with new data requires a fresh approach. That's why they introduced the idea of a 'predetermined change control plan' (PCCP).
Think of a PCCP as a blueprint you submit upfront. It tells regulators exactly how you plan to let your AI model learn and adapt over time without needing to resubmit for every minor tweak. You'll detail the kinds of changes you anticipate, the methods you'll use to make them, and the performance checks that will prove the device is still safe after each update.
This gives developers room to innovate while keeping regulators in the loop. It’s a smart shift from a one-and-done approval to a managed lifecycle that truly respects the dynamic nature of AI.
The Make-or-Break Role of Data and Bias
Your AI model is only as good as the data you feed it. In medicine, the quality of that data can literally be a matter of life and death. Finding, cleaning, and annotating high-quality training data is often the single biggest hurdle you'll face.
But it's not just about getting enough data—it's about getting the right data. Bias is a huge risk. Imagine a skin cancer detection model trained almost exclusively on images from a single demographic. It might work beautifully for that group but fail dangerously for everyone else. This isn't just a technical glitch; it's a massive patient safety and health equity problem.
You can't just code your way out of bias. Fair and effective AI comes from meticulously curating and balancing your training data to prevent real-world harm and avoid making health disparities worse.
A few strategies are non-negotiable here:
- Data Augmentation: When you can't find enough real-world examples, create synthetic data to fill the gaps for underrepresented groups.
- Fairness Audits: Make it a habit to test your model's performance across every demographic subgroup you serve.
- Transparent Reporting: Be upfront about the demographic makeup of your training data in your documentation.
Model Validation and Watching for Drift
Once your model is built, you have to prove it works reliably. This goes far beyond typical software testing. When it comes to AI in medical devices, you have to dig deep into proven strategies for reliable AI model testing to satisfy the incredibly high bar for safety and performance.
Another critical piece is Explainability (XAI). A clinician isn't going to trust a black box. If your AI recommends a course of action, the doctor needs to understand why. Building features that can explain the model's reasoning is absolutely essential for getting your device adopted in the real world.
And your job isn't done at launch. AI models can suffer from performance drift as the data they see in the wild starts to look different from the data they were trained on. You need a solid plan for continuous monitoring to catch this drift early. When it happens, you'll trigger the retraining or recalibration protocols you already laid out in your PCCP.
This AI-fueled shift isn't just happening in the clinic; it's also changing the business side of medtech. The medical device sales software market, for example, was valued at USD 1.22 billion in 2024 and is expected to hit USD 2.00 billion by 2029, climbing at a 10.8% CAGR. That growth is being pushed by AI for things like predictive sales analytics and the intense need for better regulatory tracking. A truly successful AI integration means thinking about its impact on the entire ecosystem, from development to deployment and even sales.
Answering Your Top Questions About Medical Device Software
If you're new to building medical device software, you're bound to run into questions that just don't pop up in other software worlds. Let's tackle some of the most common ones I hear from development teams and project managers. Getting these right can save you a world of headaches down the road.
IEC 62304 Versus FDA Guidance: What's the Difference?
This one trips up a lot of people. The easiest way I've found to think about it is that IEC 62304 is your "how-to" manual, while FDA guidance tells you "what to prove."
IEC 62304 is a global standard that gives you a structured lifecycle for building safe medical software. It's the playbook for planning, coding, testing, and maintaining your product in a controlled, repeatable way. The FDA, which governs the US market, looks very favorably on teams that follow this standard because it’s a strong signal that you have a solid process in place.
So, when the FDA asks for your risk analysis files, V&V results, or full traceability matrix, you're not scrambling. By following the processes laid out in IEC 62304, you're naturally generating that proof as you go. They’re really two sides of the same coin, both aimed at ensuring your device is safe and effective.
Can You Really Be Agile in a Regulated Space?
There's a stubborn myth that agile development and strict regulations can't mix. I'm here to tell you that’s just not true. Agile can work beautifully for medical devices, you just have to adapt it. We often call this approach "Regulated Agile."
The trick is to weave documentation and design controls directly into your agile sprints. Forget about a huge, panic-driven documentation phase at the very end. You build your Design History File (DHF) piece by piece, sprint by sprint.
Here’s what that looks like in the real world:
- Sprint Planning: A user story isn't just about a feature; it also includes tasks for the required documentation and risk updates.
- Definition of Done: A story isn't "done" until the code is written, it passes its tests, and all the related design control documents are updated and reviewed.
- Iterative V&V: You perform verification activities within each sprint, which builds confidence and reduces risk. Validation can then be scheduled for major milestones or before a release.
This lets your team stay flexible and get quick feedback, all while building the trail of objective evidence that regulators need to see.
Agile methodologies can absolutely thrive in a regulated environment. The secret is to make documentation and risk management an integral part of every sprint, not an activity you try to bolt on at the end.
The Biggest Mistakes I See in SaMD Development
When building Software as a Medical Device (SaMD), some mistakes are far more common—and far more damaging—than others. If you can sidestep these, you're already way ahead of the game.
The number one error is treating SaMD like a regular consumer app and completely underestimating the regulatory burden. I’ve seen teams try to slap a quality system and risk analysis on top of a nearly finished product. It's a costly, chaotic approach that is almost guaranteed to get flagged in an audit.
Another critical misstep is a fuzzy or incomplete requirements process. If you don't nail down your user needs with absolute clarity, you can't possibly verify or validate the software against them. This is a direct path to rework, blown budgets, and a product that fails to solve the real clinical problem.
Finally, putting cybersecurity on the back burner is a massive mistake. As more devices connect to networks, regulators rightfully see cybersecurity as a fundamental part of patient safety. You have to build secure design principles into your software from the very beginning. It's not an optional add-on anymore; it's a core compliance requirement.
At PYCAD, navigating these complexities is what we do best. We partner with innovators to bring cutting-edge AI to medical imaging devices, making sure every phase of development—from data management to deployment—is handled with the precision and compliance your project needs. See how we can help with your next big idea at pycad.co.





