Demystifying Medical Device Software Requirements

Medical device software plays a vital role in modern healthcare, impacting everything from diagnostics to treatment. As our reliance on this technology grows, so too does the need for a thorough understanding of its unique development requirements. These requirements aren't just regulatory boxes to check; they are crucial for ensuring patient safety and well-being.
Understanding Medical Device Software
What constitutes medical device software? It's defined as any software intended for one or more medical purposes, functioning independently of a hardware medical device. Software integrated into a device, such as the control system for a robotic surgical arm, is considered part of the device itself. However, an application used to analyze medical images from a separate scanning machine falls under the classification of medical device software. This distinction is important because it affects the applicable regulations and development processes.
This market is expanding rapidly, fueled by the demand for innovative healthcare solutions. By 2023, the medical device software market reached $24.29 billion. Projections indicate continued growth to $30.4 billion by 2024, representing a 25.2% compound annual growth rate (CAGR). Learn more about these market statistics. This growth underscores the increasing significance of software in delivering effective medical care.
Software Classification and Development Approach
The classification of medical device software is a critical factor in its development. Software is categorized based on the potential risk to patients if it malfunctions. Lower-risk software, like that used for administrative tasks, has fewer requirements. However, software directly impacting patient safety, such as that controlling drug delivery systems, demands much more stringent development and testing. Understanding the intended use and potential risks of the software is essential from the outset.
Challenges of Traditional Software Development Methodologies
Traditional software development methodologies, while often effective in other contexts, can be inadequate for medical device software. These methods may prioritize speed and flexibility. However, medical device software requires a meticulous focus on safety, reliability, and traceability. A software defect in a medical setting could have serious consequences. Specialized frameworks, including rigorous documentation, testing, and risk management, are necessary to address the unique challenges of this field.
Adapting to the Evolving Landscape
The medical device software landscape is in constant flux, with new technologies and regulations continually emerging. Companies that thrive in this environment are those that embrace change. They stay up-to-date on industry best practices and regulatory guidelines, investing in team training and robust quality management systems. This proactive approach allows them to develop and deploy innovative, safe, and compliant medical software solutions.
Navigating the Regulatory Maze With Confidence

Developing medical device software is a multifaceted process. It's not just about creating functional code; it's also about navigating a complex regulatory landscape. This intricate web of requirements can often seem overwhelming. However, companies that successfully navigate these regulations gain a significant competitive advantage. This section will explore how to turn these potential roadblocks into opportunities.
Understanding the Global Regulatory Landscape
Medical device software requirements differ around the world. These include frameworks like the FDA regulations in the United States, the EU MDR in Europe, and various other international standards. These regulations are in place to ensure both patient safety and device efficacy. They essentially provide a roadmap for developing robust and dependable software.
Documentation: A Cornerstone of Compliance
Thorough documentation is essential. It's not just a regulatory requirement; it's a fundamental aspect of good software development practice. Detailed documentation helps in tracking changes, identifying potential problems, and understanding the complexities of the system. This becomes especially crucial with medical device software where meticulous record-keeping is paramount for patient safety and legal compliance.
Regulatory requirements for medical device software registration necessitate detailed documentation. For instance, manufacturers need to provide a software description document. This document should include basic information, safety class, architecture, and runtime environment details. The safety class, which ranges from Class A (no possibility of injury) to Class C (potential for serious injury or death), is determined by the potential harm. This documentation, along with clinical evaluation documents and product instructions, helps ensure that the software meets regulatory standards and is safe for its intended use. Learn more about these data requirements.
To further illustrate the different safety classifications and their requirements, let's look at the following table:
Software Safety Classification Categories: This table compares different software safety classifications and their associated requirements.
| Safety Classification | Risk Level | Potential Harm | Documentation Requirements | Example Applications |
|---|---|---|---|---|
| Class A | Low | No injury possible | Basic software description, architecture overview | Blood pressure monitor data management software |
| Class B | Moderate | Minor injury possible | Detailed software description, risk analysis, verification and validation documentation | Insulin pump control software with limited dosage adjustments |
| Class C | High | Serious injury or death possible | Comprehensive software description, extensive risk analysis, rigorous testing and validation documentation, clinical evaluation report | Pacemaker control software |
As you can see, the level of documentation and scrutiny increases with the potential risk associated with the software. This tiered approach ensures that software critical to patient safety undergoes the most rigorous evaluation.
Device Classification and Regulatory Pathways
The classification of a medical device significantly influences its regulatory pathway. A Class I device, such as a tongue depressor, faces fewer regulatory hurdles than a Class III device, like a pacemaker. This holds true for software as well. Medical device software integrated into life-critical applications will have more stringent requirements than software used for administrative tasks. Understanding your device’s classification is crucial for developing an effective regulatory strategy.
Strategic Preparation for Regulatory Submissions
Regulatory submissions require significant time and resources. Strategic preparation can streamline this process. This preparation includes early engagement with regulatory bodies, meticulous planning, and robust testing protocols. For instance, pre-submission meetings with regulatory agencies allow for early feedback and identify potential issues, expediting the approval process.
Adapting to Regulatory Changes
The regulatory landscape is dynamic. Companies must stay abreast of the latest updates and adapt their development processes accordingly. This involves establishing a system for monitoring regulatory updates and implementing internal procedures to integrate new requirements. Think of it like updating your software: you need a structured process to manage updates without impacting existing functionality. Building this flexibility into your development lifecycle allows you to navigate regulatory changes and maintain compliance while staying on schedule. This proactive approach not only ensures patient safety, but also positions companies for lasting success in the dynamic medical device software market.
Mastering IEC 62304: Your Blueprint for Compliance

Developing medical device software successfully requires a thorough understanding of IEC 62304. This internationally recognized standard for medical device software life cycle processes provides a framework for developers to create safe and reliable software, mitigating risks to patients. Let's explore why this standard is so highly regarded and how it can benefit your development process.
Why IEC 62304 Matters
IEC 62304 is more than just a set of regulations; it's a guide to building high-quality medical device software. It directly addresses the specific challenges of this field, such as risk management, documentation, and configuration management. By adhering to IEC 62304, companies demonstrate a commitment to patient safety and foster trust with both regulators and users.
Adopting IEC 62304 can also lead to more efficient development processes. The standard's structured approach helps teams identify potential issues early, reducing costly rework and delays. This efficiency contributes to a faster time to market.
Structuring Your Development Lifecycle for Compliance
IEC 62304 emphasizes a risk-based approach, classifying software based on its potential to harm patients. Class A software presents no risk of injury, while Class C software could lead to death or serious injury. This classification dictates the required rigor throughout the software development lifecycle.
The lifecycle defined by the standard includes planning, requirements analysis, design, implementation, testing, and maintenance. Each stage has specific requirements for documentation and risk assessment, ensuring traceability and accountability. This structure ensures safety is built into every step. IEC 62304 plays a crucial role in managing the entire lifecycle of medical device software, working in conjunction with ISO 14971 (risk management) and ISO 13485 (quality management systems). Learn more about how these standards work together.
Integrating Risk Management Throughout Development
Risk management is an ongoing process integrated throughout the software development lifecycle under IEC 62304. This involves identifying potential hazards, assessing their risks, and implementing mitigation strategies. A software failure in a device like an insulin pump, for example, could have severe consequences, making robust risk control measures critical.
Traceable Documentation and Configuration Management
IEC 62304 mandates meticulous documentation. This provides a clear audit trail, demonstrating how risks were identified and addressed. This fulfills regulatory requirements and facilitates internal communication and knowledge transfer, ultimately contributing to product quality and patient safety.
The standard also emphasizes configuration management, controlling and documenting software changes. This ensures all modifications are tested and validated, minimizing the risk of introducing new errors during updates and maintenance. Configuration management helps maintain software safety and reliability throughout its lifecycle.
Real-World Implementation Insights
Companies with successful IEC 62304 implementations often highlight the importance of early planning and collaboration. Early engagement with regulatory experts can prevent costly mistakes and delays. Cultivating a culture of quality and safety within the development team is also vital for embedding the principles of IEC 62304 into the development process. These practical insights make the standard a valuable resource for medical device software developers. By mastering IEC 62304, your company can achieve compliance and build better, safer software that improves patient outcomes.
Conquering SaMD Challenges: Strategy for Stand-Alone Success

Software as a Medical Device (SaMD) is a rapidly expanding field in healthcare technology. Developing this type of software requires a specialized approach because of unique regulatory pathways and inherent challenges. This section explores strategies successful companies use to navigate these complexities.
Understanding the Unique Regulatory Landscape for SaMD
Unlike software embedded in a hardware medical device, SaMD functions independently. This stand-alone operation requires specific regulatory considerations. It's essential to understand that SaMD, even without hardware, is classified as a medical device. This classification significantly impacts development, testing, and deployment.
For example, a mobile app diagnosing skin conditions based on uploaded images is considered SaMD. As such, it's subject to specific medical device software requirements. SaMD's independent operation, while distinct from hardware devices, doesn't exempt it from medical device regulations. The FDA offers specific SaMD guidance, emphasizing validation to ensure safety and effectiveness, a crucial aspect of patient safety.
A malfunctioning diagnostic software could have serious health consequences, highlighting the need for thorough validation. Learn more about SaMD.
Effective Strategies for Cloud-Based and Mobile Health SaMD
Cloud-based and mobile health represent key growth areas for SaMD. They also introduce unique development challenges. These include ensuring data security, maintaining HIPAA compliance, and addressing cybersecurity vulnerabilities.
Successful companies prioritize these concerns. They implement robust security measures, encrypt data, and conduct regular security audits. These practices protect patient information and maintain regulatory compliance in increasingly interconnected environments.
Addressing Data Privacy Requirements for SaMD
Data privacy is paramount in healthcare. SaMD developers must address strict regulations like HIPAA, GDPR, and other international standards. A practical approach involves incorporating data privacy considerations from the design phase. This might include implementing data anonymization techniques, limiting data collection to necessary information, and obtaining informed consent from users.
These measures demonstrate regulatory compliance and build trust with patients and healthcare providers.
The FDA's Digital Health Software Precertification Program and SaMD
The FDA’s Digital Health Software Precertification (Pre-Cert) Program aims to streamline regulatory pathways for SaMD. This program focuses on evaluating the software developer’s quality management system rather than individual products.
This approach promotes innovation while upholding rigorous safety standards. It allows companies with proven track records to bring products to market faster. Understanding and participating in the Pre-Cert program can be a strategic advantage for SaMD developers in this evolving regulatory landscape.
Positioning Your SaMD for Market Success
Launching a SaMD product successfully requires more than just meeting regulatory requirements. A comprehensive market strategy is essential. This involves understanding the target audience, identifying market needs, and developing a compelling value proposition.
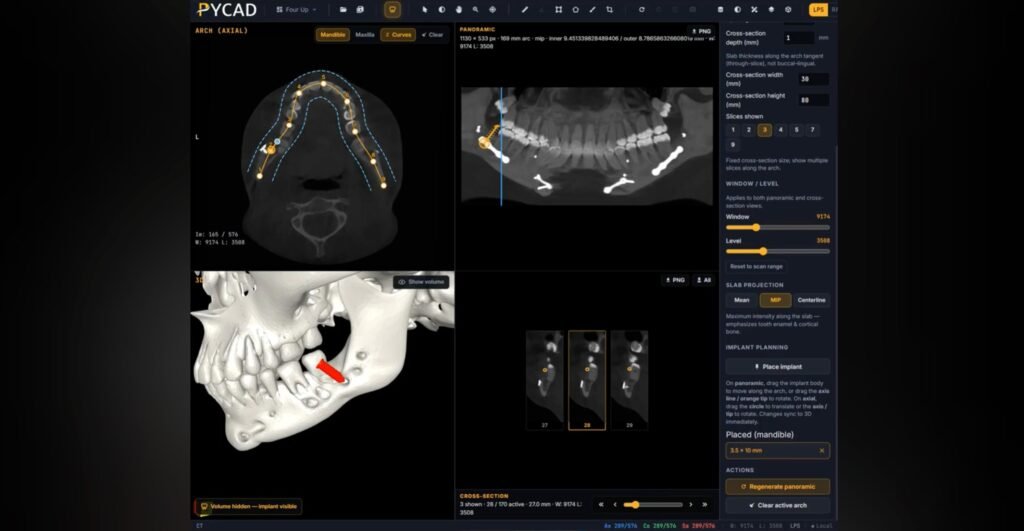
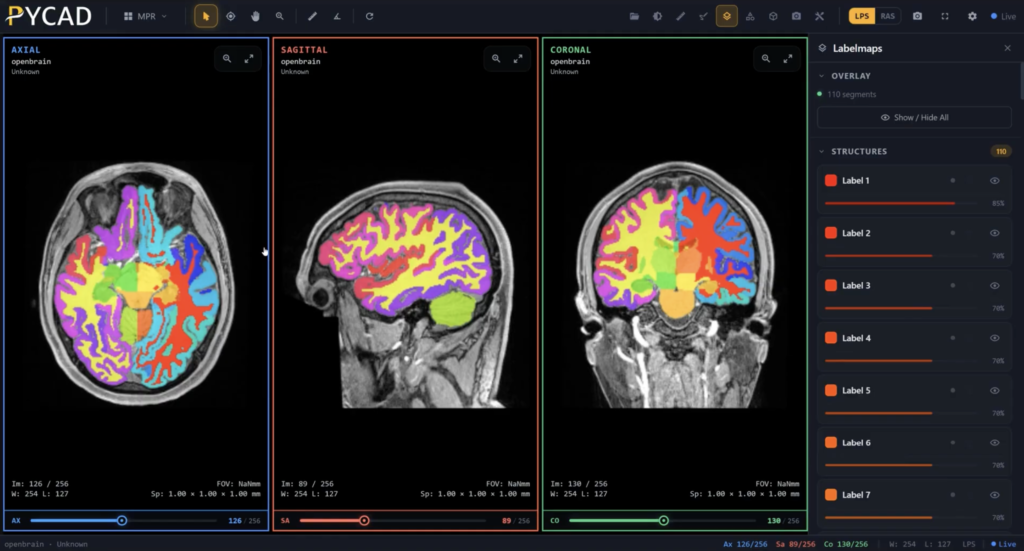
For example, PYCAD specializes in integrating AI into medical imaging solutions. Our expertise spans data handling, model training, and deployment, helping clients improve diagnostic accuracy and operational efficiency. Discover more about PYCAD's AI solutions. By combining regulatory compliance with a strong market focus, SaMD developers can position their products for success and contribute meaningfully to healthcare technology advancements.
Validation Strategies That Actually Work
Validating medical device software isn't just about checking regulatory boxes; it's about ensuring patient safety and product effectiveness. This section moves beyond the theoretical and dives into practical validation strategies that meet regulatory requirements without overburdening development teams.
Establishing Meaningful Acceptance Criteria
Effective validation starts with well-defined acceptance criteria. These criteria outline the conditions that the software must meet to be considered validated. They need to be objective, measurable, and directly related to the software's intended use and risk assessment.
For example, software controlling an insulin pump might have an acceptance criterion like, "The delivered dose must be within ±0.1 units of the prescribed dose." This specificity eliminates ambiguity and provides clear direction for testing.
Implementing Appropriate Testing Methodologies
Choosing the right testing methodologies is essential for comprehensive validation. Different software functions and risk levels require tailored testing approaches. Unit testing, for example, verifies individual software components.
Integration testing checks how these components interact, while system testing evaluates the entire software system. User acceptance testing validates the software from the end-user's point of view.
To better understand these methodologies, let's look at a comparison table. This table breaks down the purpose, timing, documentation, and regulatory considerations for each testing method.
Medical Device Software Testing Methodologies: This table compares different testing methodologies.
| Testing Method | Purpose | When to Use | Documentation Required | Regulatory Considerations |
|---|---|---|---|---|
| Unit Testing | Verify individual software components | During development | Unit test plans, test cases, results | Ensures each component functions correctly |
| Integration Testing | Assess interactions between software components | After unit testing | Integration test plans, test cases, results | Verifies proper communication between components |
| System Testing | Evaluate the complete software system | After integration testing | System test plans, test cases, results | Ensures overall system functionality and performance |
| User Acceptance Testing | Validate software from the end-user perspective | Before release | User acceptance test plans, test cases, results | Confirms the software meets user needs and intended use |
This targeted testing approach optimizes resource allocation and focuses efforts on critical areas. It also makes it easier to create traceable documentation, which is essential for regulatory compliance.
Handling Test Failures Effectively
Test failures are a normal part of software development. They offer valuable opportunities to learn and improve. A robust validation strategy includes a process for documenting, analyzing, and resolving these failures.
For example, a root cause analysis helps identify the underlying reasons for failures. This allows developers to implement corrective actions and prevent recurrence. This iterative process strengthens the software and builds confidence in its reliability. Documenting test failures and the corrective actions taken demonstrates a commitment to quality, which is important for regulatory compliance.
Adapting Validation to Different Development Phases
Validation should be integrated throughout the software development lifecycle. During the requirements phase, validation focuses on ensuring requirements are clear, complete, and testable. In the design phase, validation ensures the design meets the requirements and addresses potential risks.
During implementation, testing verifies that the code functions as expected. Finally, post-market surveillance monitors software performance and identifies any unforeseen problems. This continuous quality control minimizes late-stage issues and ensures the software consistently meets requirements. This proactive approach saves time and resources. PYCAD supports clients through this validation process, offering comprehensive services, including data handling, model training, and deployment, ensuring adherence to medical device software requirements. This allows development teams to focus on innovation while remaining compliant.
From Development to Market: Breaking Through Barriers
The journey of bringing medical device software to market presents numerous challenges. Even the most groundbreaking software can face unexpected roadblocks. This section, informed by experienced professionals, examines proven strategies to successfully navigate the complexities of bringing a product from development to commercialization.
Managing Development Costs While Maintaining Compliance
One of the primary challenges in medical device software development is balancing development costs with strict regulatory requirements. This requires careful planning and resource allocation. A practical approach involves prioritizing features based on risk and clinical need. Focusing on core functionality early in the development process helps ensure efficient resource utilization and minimizes unnecessary spending on non-essential features for initial regulatory approval.
Accelerating Approval Timelines Without Compromising Quality
Speed to market is paramount in the competitive medical device software industry. However, rushing development can jeopardize quality and lead to regulatory setbacks. A structured, Agile development approach, incorporating quality checks at each stage, can facilitate rapid iteration while upholding necessary standards.
Building an Effective Regulatory Strategy From Day One
Regulatory strategy should be an integral part of the development process, not an afterthought. Early and consistent communication with regulatory bodies, like the FDA, is vital. This proactive communication helps identify potential issues early, preventing costly revisions later.
Navigating this complex landscape is crucial. Medical device development and commercialization face substantial regulatory hurdles. In 2020, the FDA received over 3,000 applications via the 510(k) pathway, a common route for medical technology approvals. Yet, many inventions never reach the market due to regulatory complexities and substantial development costs, which can range from $31 million to $94 million. The limited availability of regulatory expertise within academic settings further complicates these challenges. Explore this topic further here.
Implementing Post-Market Surveillance That Informs Future Development
Post-market surveillance is more than a regulatory requirement; it's a valuable opportunity to collect real-world data on software performance and user experience. Successful companies use this data to shape future development, identify areas for improvement, and proactively address emerging issues. This creates a continuous feedback loop for enhancing product quality, safety, and effectiveness.
Avoiding Common Pitfalls That Lead to Regulatory Delays
Several common pitfalls can hinder medical device software development, leading to regulatory delays. These include insufficient documentation, inadequate testing, and poor communication with regulatory bodies. Proactive risk management, comprehensive testing protocols, and meticulous record-keeping can mitigate these risks and ensure development stays on track.
PYCAD: Your Partner in Navigating Medical Device Software Requirements
PYCAD specializes in integrating AI into medical imaging solutions, providing support throughout the entire development lifecycle. From data management and model training to deployment and regulatory compliance, PYCAD helps medical device software companies navigate the complex path to market. PYCAD’s expertise ensures products meet the highest quality, safety, and efficacy standards. Learn more about how PYCAD can help your medical device software project succeed at https://pycad.co.